
 Andre Moa
Andre Moa
 6 Dec 2019
6 Dec 2019
 10 minute read
10 minute read

Launch excellence for pharmaceuticals depends on a multitude of factors, from pricing, reimbursement and health technology assessment (HTA) to positioning, sequencing, sales, marketing, media coverage and the competitive landscape. None of these can get off the ground, though, without regulatory approvals in key markets. Here, we tally up 2019 new drug approvals to date in the US and the European Union.
The number of new drugs and biologics authorised centrally in the year to date by the US Food and Drug Administration and the European Medicines Agency, the two most influential agencies for companies’ global drug launch programmes, looks unlikely to match the record achieved in 2019. Nonetheless, it reflects the continuing trends of diversified, boundary-pushing innovation, and of speciality drugs targeting more precisely segmented patient populations, that have driven pharmaceutical market growth in recent years.
In summary, as of mid-November 2019 the US FDA had approved a total of 38 new medicines and therapeutic biologics (through CDER) as well as 22 new biological agents (through CBER). The FDA approvals 2019 count to date compared with 59 new medicines/therapeutic biologics (CDER) and 39 biological agents (CBER) authorised for marketing in 2018. The EMA’s Committee for Medicinal Products for Human Use had issued positive opinions on 39 new medicines, therapeutic biologics and vaccines by the middle of November 2019. It recommended 53 such products for approval in 2018.
Like-to-like approval comparisons are complicated
Like-to-like comparisons of new drug and biologic approvals by the FDA and the EMA are clouded by varying definitions of what constitutes innovation and what is actually ‘new’. On a more simplistic level, the EMA grants centralised marketing authorisations, valid for the whole of the European Union, to chemically based medicines as well as biologicals and vaccines. In the US, these approvals are split between two FDA divisions, the Center for Drug Evaluation and Research (CDER) and the Center for Biologics Evaluation and Research (CBER).
A further muddying factor is that the CDER lists approvals of 'novel medicines', which are typically (but not exclusively) new molecular entities or NMEs. Those approvals include diagnostic agents (e.g., in 2019, IQ Medical Ventures’ ExEm Foam (air polymer-type A)) but not vaccines and gene therapies approved by the CBER and falling, in the EU, under the EMA umbrella. The CDER approves therapeutic biologics and well as chemically based medicines, but not vaccines, allergenic products, blood/blood products, plasma derivatives, or cellular/gene therapy products. All of these latter go through the CBER, although many products approved under a Biologics License Application in the US are screening tests, assays or reagents.
Final EU approvals can be slow to emerge
The EMA submits ‘new medicines’ for assessment by its Committee for Medicinal Products for Human Use (CHMP), whose positive opinions are subject to final sign-off by the European Commission. Only then are the products effectively ‘EMA approved’. FDA approvals are also on the recommendation of its various advisory committees, but final approvals are listed on the agency’s website.
There may be several months’ lag between a positive CHMP recommendation and European Commission grant of a marketing authorisation. Moreover, whether ‘new medicines’ constitute ‘new active substances’ (NASs) – the nearest equivalent to FDA-approved NMEs in the US – is not determined (or rather publicly available) until the Commission has signed off the approval and the relevant marketing-authorisation documentation has appeared on the EU’s website.
That not all CHMP-recommended new medicines are NASs is evident from, for example, the EMA’s listing of Acorda Therapeutics’ Inbrija in July 2019 as a recommended new medicine for the symptomatic treatment of ‘off’ periods in Parkinson’s disease. In fact, this is an inhaled powder formulation of levodopa, which has long been a staple treatment among the limited range of therapeutic options available for Parkinson’s.
FDA and EMA approvals are largely in tune
A more meaningful comparison is therefore year-to-year approvals by the EMA and the FDA (CDER plus CBER authorisations), respectively. It is worth noting, however, that a recent joint EMA/FDA analysis of decisions taken by the two agencies on 107 new medicine applications between 2014 and 2016 found that more than 90% of these decisions were aligned. This partly reflected increased investment in dialogue and co-operation between the EMA and the FDA (e.g., joint working groups) since 2003.
With those provisos in mind, we can count positive recommendations by the EU’s CHMP in the year to date (noting where these have already been approved by the European Commission) and all the marketing authorisations granted so far to novel medicines, gene therapies and vaccines by the US FDA. The FDA approvals do not include generics and biosimilars.
The EMA/CHMP scores leave out generics and biosimilars, as well as products approved through hybrid or informed consent procedures that rely on pre-existing data from other medicines or biologics. At the time of writing, 25 of the 39 new products recommended for EU approval by the CHMP had secured marketing authorisations from the European Commission, according to publicly available information.
What approvals say about the evolving launch landscape
Other than the numbers, then, what do these approvals tell us about the evolving landscape for new drug launches in 2019 and beyond? In volume terms, the obvious take-out is that 2018 was an exceptional year on both sides of the Atlantic.
It should not necessarily be inferred from that, though, that the FDA or EMA have slackened off in 2019. Any number of factors can influence FDA approval or CHMP opinion tallies for drugs and biologics from one year to the next, including the quality and timing of submissions, spill-overs from late in the year, internal resources at drug agencies, and one-off factors such as the EMA’s Brexit-related transfer from London to Amsterdam.
Beyond the numbers, there has been breakthrough innovation, in the form of gene therapies such as Novartis/Avexis’ Zolgensma (onasemnogene abeparvovec) for spinal muscular atrophy; or bluebird bio’s Zynteglo (Autologous CD34+ cells encoding βA-T87Q-globin gene) for beta-thalassaemia. The roll-out into the US and EU of rare-disease gene therapies with very high price tags (e.g., $2.1 million per patient for Zolgensma in the US) and a ‘one-shot’ treatment proposition has prompted debate about whether these costs are sustainable by already hard-pressed healthcare systems, or whether creative financing mechanisms can ease the burden.
Rare-disease therapies are still riding high
Orphan or rare-disease medicines, encouraged by development incentives, scientific advances in precision medicine and limited to non-existent market competition, remain a prominent feature of the approvals picture for 2019. In the EU, for example, ten of the 39 products so far recommended for approval by the CHMP have been orphan medicines. In 2018, 21 of the 53 new medicines that received positive CHMP opinions over the course of the year were orphan-designated.
Other notable examples of innovation among the 2019 approvals crop include Bayer/Loxo Oncology’s Vitrakvi (larotrectinib), the first time an exclusively ‘tumour-agnostic’ (rather than site-specific – e.g., breast, lung or prostate) cancer drug has secured marketing authorisations; and Epidyolex (cannabidiol) for the treatment of seizures associated with Lennox-Gastaut syndrome or Dravet syndrome. This was the first time a medicine with an active substance derived from cannabis had gained EMA approval through the agency’s centralised procedure.
And potential blockbusters keep coming
Of course, from a commercial perspective – and, indeed, in the interests of patient access to the safest, most effective medicines – all of that innovation is only meaningful if the product launches successfully, is priced for value, and is taken up (and paid for) by healthcare systems. For companies with innovation to market, pharmaceutical launch success inevitably comes down to revenues, competitive edge, profit margins and lifecycle management.
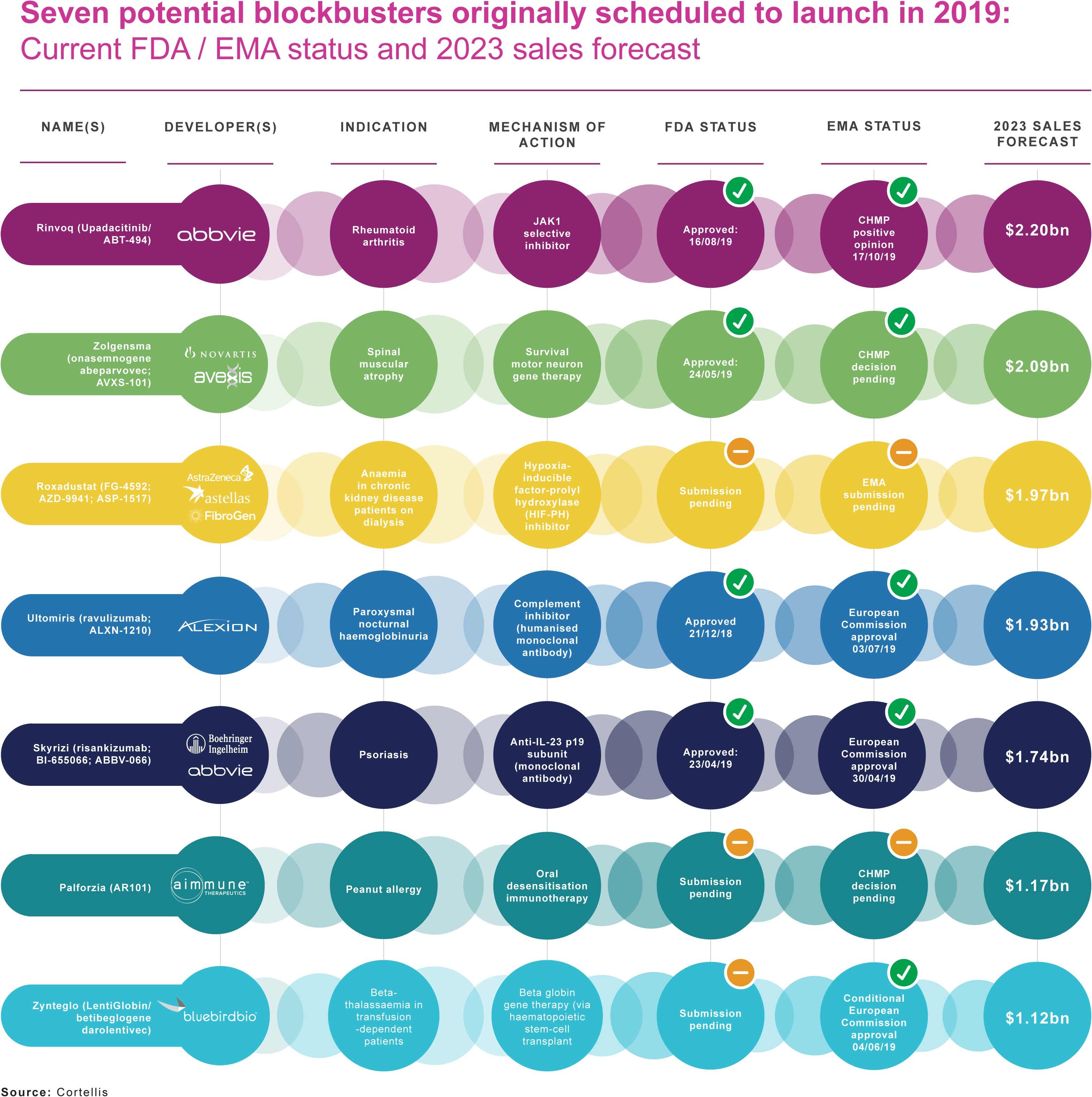
Among new drugs reported as launched or soon to launch in 2019, commentators have singled out a number of potential blockbusters that testify to the continued potency both of pharma R&D and of launch excellence platforms. These include upadacitinib, AbbVie’s JAK1 inhibitor for rheumatoid arthritis; Alexion’s Ultomiris (ravulizumab) for the rare blood disorder paroxysmal nocturnal haemoglobinuria; Boehringer Ingelheim/AbbVie’s psoriasis treatment Skyrizi (risankizumab); Novartis/Avexis’ aforementioned gene therapy Zolgensma (onasemnogene abeparvovec) for spinal muscular atrophy; and Aimmune’s peanut-allergy immunotherapy Palforzia (AR101).
Cortellis’ Drugs to Watch report for 2019 listed the following seven drugs as set to enter the market this year and achieve blockbuster status by 2023:
Last Updated: 05th December 2019
R&D pipelines continue to expand
Looking to the future, R&D pipelines continue to expand year by year. Informa’s annual Pharmaprojects Pharma R&D review identified a total of 16,181 ongoing pipeline projects in early 2019, 6% more than in the previous year’s analysis.
With rapidly aging populations worldwide, Alzheimer’s and Parkinson’s disease remain the Holy Grail for drug developers. However, these diseases have also proved uniquely resistant to generating compelling outcomes in clinical studies, and especially in pivotal Phase 3 clinical trials. In the meantime, better targeted compounds, unmet needs and orphan-drug incentives have expanded opportunities in more established categories such as oncology.
One prominent example is Merck & Co’s cancer immunotherapy Keytruda (pembrolizumab), which is already authorised for more than 20 oncology indications in the US and is widely approved elsewhere. According to recent projections by GlobalData, Keytruda will overtake AbbVie’s Humira (adalimumab) – now under pressure from several biosimilars in Europe – as the world’s top-selling drug in 2023, generating annual sales of $22.2 billion by 2025.
Approvals are nothing without launch excellence
If approval volumes for 2019 in the US and the EU will struggle to attain last year’s peaks, there is still plenty of evidence that drug development pipelines continue to produce a rich crop of new medicines, some of them offering breakthrough innovation and some destined for blockbuster status.
For those approvals to translate into commercial success and better patient outcomes, though, companies need the right digital technology to ensure that milestones, timelines and challenges in their pharma launch plan are visible, aligned, understood and actionable in real time across different countries, functional silos and layers of management.
To cite leading (bio)pharmaceutical services provider IQVIA, launch success “starts with an informed strategy that reaches for the latest data, technology, analytics and market expertise to ensure decisions are swift and right”. In an environment where digital technology and media are disrupting the pharmaceutical model from research to market entry and beyond, the journey from approval to launch excellence must also be guided by the best available digital capabilities.
With technology-enabled cross-functional alignment and team collaboration across the broad spectrum of activities constituting pharmaceutical launch readiness, pharma and biopharma companies can harness their best expertise and resources towards the common goal of making good on this year’s drug approvals, and those to come.
TRiBECA® Knowledge provides pioneering software to some of the world’s leading pharmaceutical companies, to help them optimise launch, market access and tendering operations. Our launch readiness platform SmartLaunch™ helps pharmaceutical companies to optimise launch readiness by enhancing visibility, transparency, communication and collaboration across products, management layers, business functions and countries worldwide. This will drive better-informed decisions around launch strategy and execution, while avoiding unnecessary delays and improving speed to market, to the benefit of the business and patients alike.