
 Andre Moa
Andre Moa
 12 Dec 2023
12 Dec 2023
 19 minute read
19 minute read

Every product needs a market. It also needs market access. For pharmaceuticals, that means HTA, reimbursement, pricing, positioning and other elements that can make all the difference between a spectacular launch and a costly also-ran. In our pharma trends for 2024, we look at some critical market access challenges in key territories for industry, and how companies can best prepare for them over the coming year.
The bad news is that market access is not getting any easier. That reflects several broader trends shaping pharma, such as relentless pressure on health system finances and resources, exacerbated by COVID-19, greying populations and age-related diseases; political expediency in health funding and decision making; more complex and expensive therapies; the continuing rise of payer power; and digital windfalls such as rapid information exchange and more sophisticated analytics.
For companies really tuned in to what is coming, and how to deal with it both at global and local level, these are not just challenges but potentially crucial differentiators in a fiercely competitive marketplace. Without optimal pharma market access, launch excellence can only ever be an ambition with limited real-world relevance for companies, shareholders, health systems, healthcare professionals or patients.
Pharmaceutical Trends for 2024
Trend #1. EU's pharma review: New challenges for market access teams
If you had a new and exciting medicine, approved centrally for use in all 27 EU member states, would you not want to launch it in as many of those markets as possible and as quickly as possible, reaching the maximum number of patients? Theoretically yes; in practice, not necessarily.
Patient access and market access sometimes have to part company. Of course, patient access is fundamental to market access for a new launch. But Market Access is also about optimising commercial potential. That means launching at the right time, and with the right pricing, reimbursement and value proposition. The flexibility to do so, in a nominally harmonised EU market, runs against the grain of new proposals to square market and patient access across Europe.
In reality, pricing, reimbursement and HTA procedures are pharmaceutical industry trends subject to sharp variations, both globally and within the EU. Available funding for new drugs depends partly on healthcare priorities and local economies. HTA capabilities may be more evolved in some countries than others, with different emphases in their assessment criteria (budget impact, cost-benefit, etc). Pricing and reimbursement systems have their own methodologies, requirements and timelines.
That significantly complicates pan-EU launches and their sequencing. Launch programming must also take into account the potential for price referencing from one member state to another and for parallel importing from lower to higher-priced markets. Accordingly, a company may want to prioritise launches in member states with more permissive pricing and reimbursement.
It may even choose to delay or forego launch in some markets, if available pricing and reimbursement are not only uneconomical but liable to undermine market entry in other member states. This has happened several times in Germany (traditionally a go-to-market) recently, and particularly with new cancer drugs (see below).
It partly reflects legislative changes in Germany that have reined in free pricing and yoked drug reimbursement more strictly to added value. The trend is also symptomatic of an HTA system that has rejected progression-free survival as a surrogate endpoint worthy of added-benefit ratings in oncology.
The EU’s pharmaceutical review
This is the context for perhaps the most contentious of the proposals in the European Commission’s sweeping review of EU pharmaceutical legislation, published on 26 April 2023 and now before the European Parliament. The Commission wants to reduce baseline regulatory data protection (RDP) for new medicines by two years to six years, plus two more years of market exclusivity.
The lost RDP would be added back if medicines were made available for continuous supply in all member states within two years of approval. The protection period could also be extended if drugs met other criteria, such as addressing unmet medical needs or conducting comparative clinical trials (both restoring six months’ RDP).
This more coercive stance reflects a core objective in the Commission’s pharmaceutical review: making sure all patients have timely and equitable access to safe, effective and affordable medicines. Yet the carrot-and-stick approach immediately raises the question of whether pan-EU launches within two years are even achievable, given that they depend partly on the discretion and resources of individual member states.
In some of these countries, pricing and reimbursement procedures can certainly drag. The European Federation of Pharmaceutical Industries and Associations’ (EFPIA) latest Patient WAIT Indicator found that, on average, new medicines were reaching patients within 128 days of centralised approval in Germany, 436 days in Italy, 629 in Spain, 918 in Romania and 1,351 in Malta. The EU average time to availability in 2018-201 was 517 days.
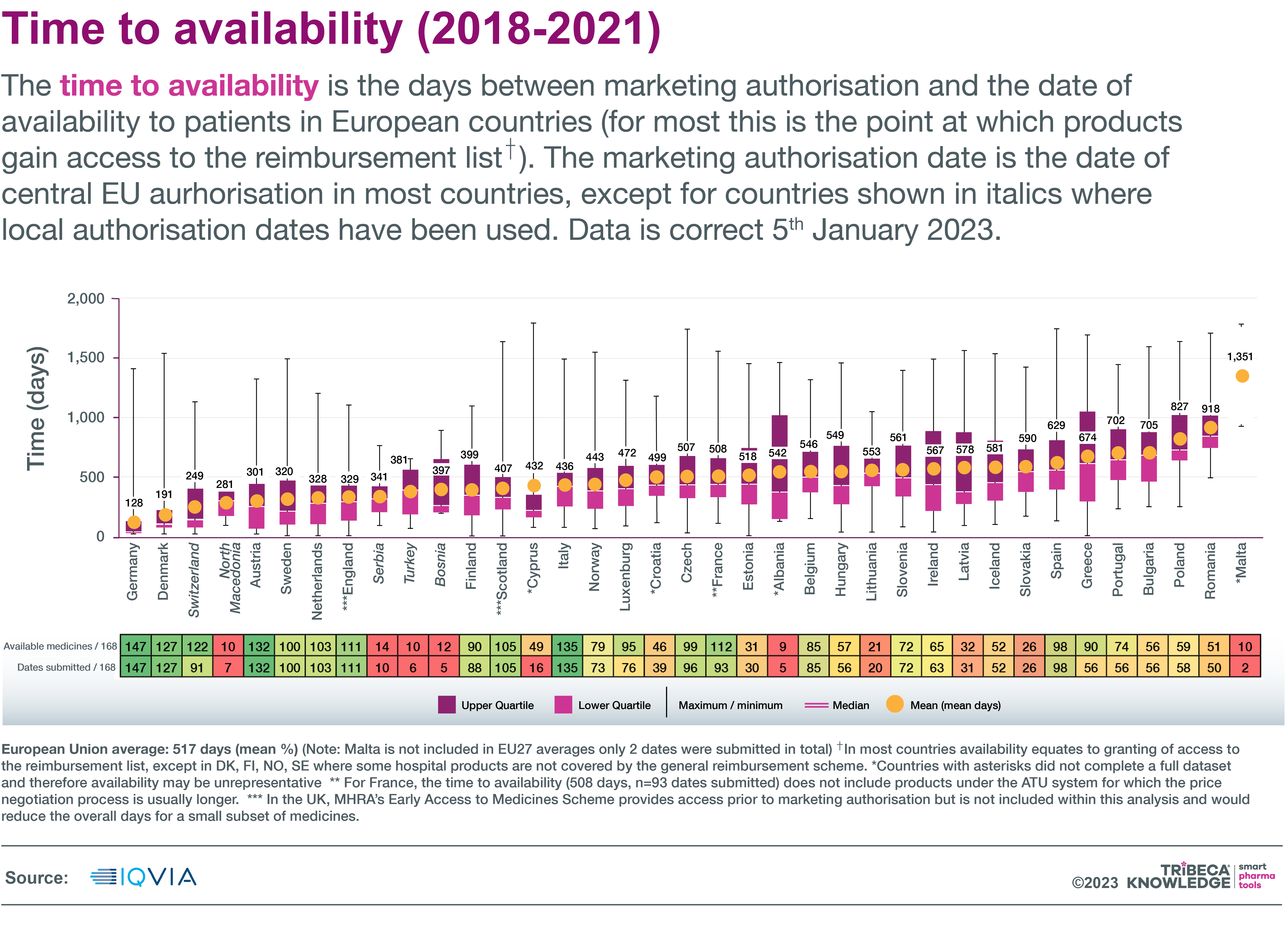
Source: IQVIA
Why delays occur
More recent data from the industry-led European Access Hurdles Portal suggest that only about one-quarter of the lag in patient availability is due to delays in company submissions. The remainder is down to hold-ups in national pricing and reimbursement processes, EFPIA says.
Even tardy market access submissions are for different reasons: mostly value-assessment processes and evidence requirements in Western Europe, whereas in Eastern Europe delays are more about health-system constraints and their impact on corporate decision-making. “This shows that there is no one-size-fits-all-solution to fix access issues, and regional or country-specific solutions are needed,” EFPIA states.
There are also fears that the two-year launch window might result in drug supply problems or that, with the clock ticking, companies might be pressured into accepting unfavourable pricing or reimbursement in some markets as a condition of entry. Furthermore, EFPIA estimates that reduced baseline RDP would cut the incentive for companies to invest in breakthrough medicines in Europe by 55% over the next 15 years. This is at a time when the region’s share of global R&D investment is predicted to fall from 32% to 21% by 2040.
More encouraging provisions
There are more innovation-friendly provisions for the biopharma industry in the pharmaceutical package, such as reducing the time taken to evaluate and sign off new medicines centrally in the EU. There are also plans for regulatory ‘sandboxes’, with adaptive frameworks including real-world data collection, to expedite approvals of novel therapies.
Shorter RDP periods are not industry’s only concern, though. EFPIA has questioned, for example, the high bar for meeting unmet or high unmet medical need to earn additional RDP or orphan-market exclusivity; and more stringent requirements for timely orphan drug development.
As things stand, the revisions may be some distance from implementation. The pharmaceutical package already had a late start in 2023, while the chances of the European Parliament and Council completing their discussions before parliamentary elections in 2024 look slim. The issue is further complicated by emerging differences between member states on issues such as the right balance between patient access to medicines and incentives for innovation.
There may also be significant changes to come. A recent report on the Commission’s proposals by parliamentary rapporteur Pernille Weiss recommended, for example, a longer baseline RDP period (eight to nine years), coupled with adjustments to the timely-launch requirement and changes to the definition of unmet medical need qualifying for extended RDP.
Whatever the outcome of these deliberations, they could set the tone for market access trends in 2024 and make the European Union a key battleground for launch excellence strategies that satisfy commercial and patient interests without undermining either.

Trend #2. Price pressures intensify: Anything the IRA can do, Germany and Japan can do better?
Drug-pricing provisions in last year’s Inflation Reduction Act marked a significant departure from the free-pricing model that made the US a premier launch market for new medicines. The IRA gave the Centers for Medicare and Medicaid Services authority to negotiate Maximum Fair Prices (MFPs) for high-cost drugs used in the Medicare scheme. It also included rebate penalties for companies that raised Medicare drug prices above inflation.
Among the more taxing current trends in the pharmaceutical industry, multinationals used to grappling with complex and variable price controls in key markets like Europe or Japan may have felt the view from the other side of the Atlantic or Pacific did not look so bad after all. The pricing environment in Europe and Japan is only getting tougher.
MFPs kick in
The first batch of 10 Part D medicines subject to MFPs, including blockbusters like Bristol-Myers Squibb’s Eliquis (apixaban) and Eli Lilly’s Jardiance (empagliflozin), was announced in August 2023. The negotiated MFPs will come into effect on 1 January 2026. Minimum discounts applied to MFP-eligible medicines will range from 25% to 60%, depending on how long a drug has been on the market. Penalties for inflation-busting price increases are already well in hand, with second-round rebates on 43 drugs announced in June 2023.
The broader impact of these controls on US market access, commercial strategies and drug development remains uncertain. It will depend partly on factors such as how much an asset is exposed to the Medicare market, and any potential spillover into, or trade-offs with, commercial-market segments.
There may even be worse to come for the pharma industry. President Biden’s federal budget proposals for the fiscal year 2024 include plans to double the number of drugs subject to Medicare price negotiation, reduce and equalise grace periods for both biologics and small-molecule drugs, and extend penalties for above-inflation price increases to the commercial sector.
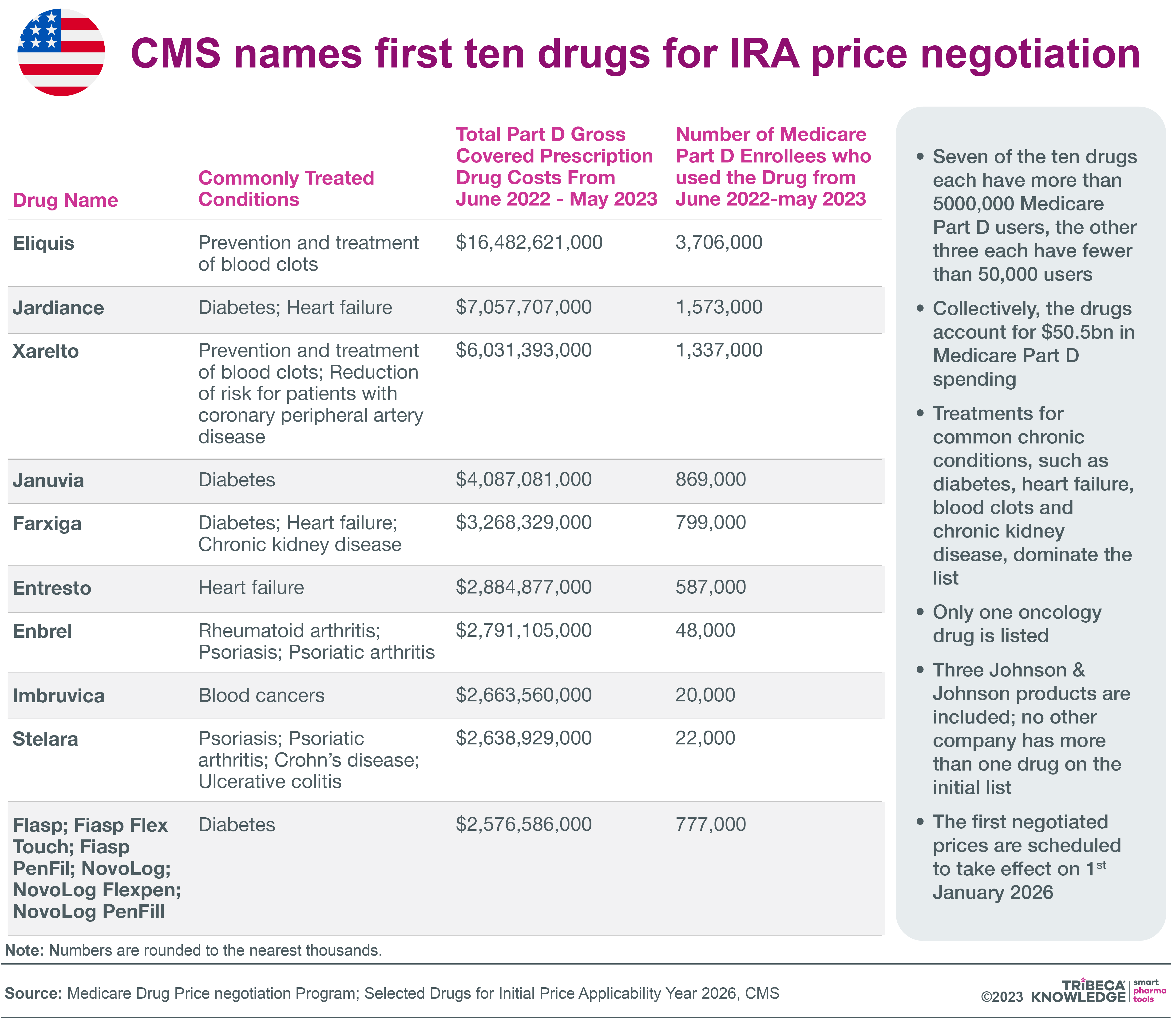
Source: Neil Grubert
Cold climate in Germany
Further afield, a recent development with implications for drug pricing across the EU is the Commission’s proposals for wide-ranging pharmaceutical reform (see above). These do not address drug pricing directly; that remains, along with reimbursement policies, a national responsibility. However, the proposals do risk undermining launch strategies driven by price discrepancies across the region and their domino effects on reference pricing or parallel importing.
For example, the Commission wants to cut regulatory data protection and market-exclusivity periods for new medicines, unless they are launched in all member states within two years of approval. This should improve equity of access for patients but could also dilute preferential launch strategies geared to traditionally higher-priced markets, such as Germany. That said, even the German market is starting to lose its appeal.
The Act on the Financial Stabilisation of the Statutory Health Insurance System (GKV-FinStG), which took effect in November 2022, includes measures such as:
- Reducing the free-pricing period for newly launched drugs from 12 to six months
- Lowering to €30 million the annual turnover threshold above which orphan drugs are subject to full benefit assessments
- A one-year increase to 12% of sales in the mandatory discount to statutory health insurers
- A mandatory 20% rebate on new combination therapies
- Stricter pricing ‘guardrails’ for medicines without significant added benefit: for example, the reimbursement price of a drug with no added benefit must be at least 10 percent below that of the most economical non-generic comparator.
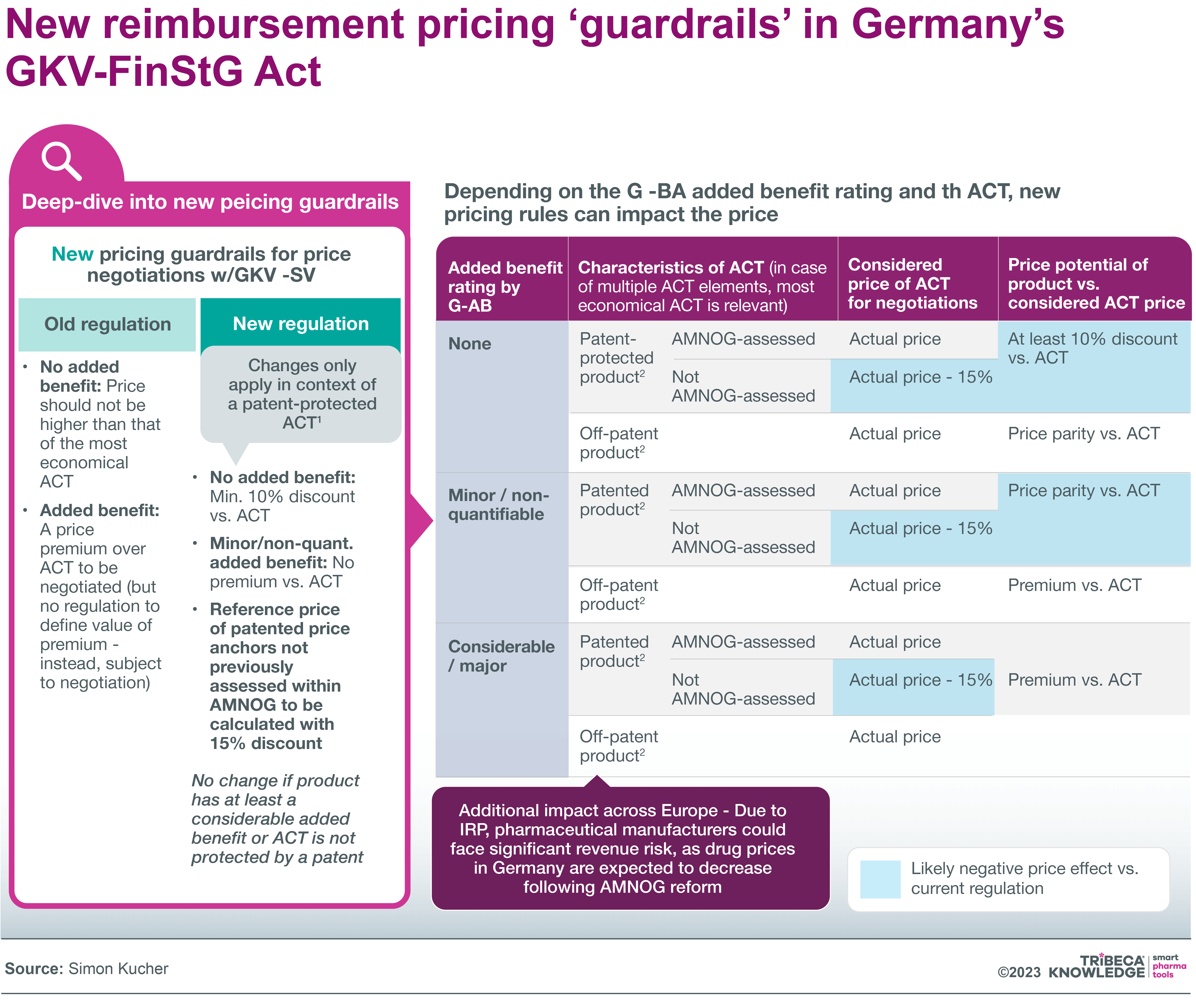 Source: Simon Kucher
Source: Simon Kucher
The new pricing and reimbursement provisions have been blamed for a number of decisions not to launch or to withdraw drugs from the market. These include Bristol-Myers Squibb’s anticancer Opdualag (nivolumab + relatlimab); Boehringer Ingelheim’s Spevigo (spesolimab) for psoriasis flares; and Novartis’ lung-cancer drug Tabrecta (capmatinib). A recent survey by research-based association VFA found that 21 of 48 member companies had decided to delay or avoid launching new drugs/indications in Germany, or were considering doing so, as a direct result of the GKV-FinStG measures.
Japan doubles down on price cuts
Japan is another important market in which pharma is feeling the pricing pinch. Additional ‘off-year’ price revisions for pharmaceuticals, over and above Japan’s regular biennial reviews, were implemented in 2021. They re-emerged in 2023. The latest off-year revisions took effect on 1 April 2023, applying to drugs with discrepancies between market prices and National Health Insurance prices exceeding 0.625 times the average price variance rate in 2022
This led to price reductions on more than 2,000 medicines in April, with the worst-hit patent-protected drugs weathering average price cuts of 8-9%, according to GlobalData. There were also some price increases, as special measures were introduced to ensure stable supply of designated drugs. This meant price premiums for 143 innovative or unprofitable medicines benefiting from price premiums, while another 90 drugs held their prices under the regular price-maintenance premium programme.
Otherwise, 240 products saw their prices increase in the April revision, by an average of nearly 29%. Despite these gains, though, prices for 91% of medicines subject to price revision in Japan were either unchanged or cut, GlobalData noted. It further warned that special pricing measures for unprofitable drugs were unlikely to continue during fiscal year 2024, which would “only heighten the already strong criticism by industry representatives of the government on the frequency of price adjustments”
An expert panel at the Ministry of Health, Labour and Welfare echoed those concerns in a draft report earlier this year. With Japan losing its edge as a global innovator in recent years, industry should be encouraged to take risks, the panel suggested. That required fresh thinking on critical aspects of the drug-pricing system, such as maintaining prices while new medicines were under patent, instead of just wearing them down with annual price revisions.

Trend #3. Getting ready for EU HTAR: It’s crunch time…
As of December 2023, pharmaceutical companies have little more than a year left before significant changes occur in the European landscape for health technology assessments. That makes 2024 a critical year of adjustment and creative thinking for market access teams sensitive to leading pharma industry trends and the vagaries of EU market access.
There are still unanswered questions about how much the European Union’s Regulation (EU) 2021/2282 on health technology assessment (HTAR), in force since January 2022, will achieve in harmonising facets of HTA across Europe. It is also debatable whether that harmonisation will benefit pharmaceutical companies looking for more seamless technology assessments that trickle down to the national level.
Companies will lose nothing, though, by ensuring they are as informed and prepared as possible to absorb the regulation’s impact. As consultants Deloitte warn, there is “an urgent need for pharmaceutical and MedTech companies to rethink the way they have historically organised themselves internally and prepared for country-specific HTA submissions”.
The new regulation establishes a legal and organizational framework for co-operation between member states on EU-level joint clinical assessments (JCAs) of new or existing health technologies. The JCA process starts with novel cancer drugs and advanced therapy medicinal products in January 2025, expanding to orphan drugs from 13 January 2028. All other medicines approved centrally through the EMA will be subject to JCAs from 13 January 2030.
Overseeing all of this will be a centralised Coordination Group with representatives from national HTA authorities and input from a stakeholder network including patient, healthcare professional and industry associations, as well as clinical/scientific societies and consumer organisations. The HTA Regulation also creates a framework for joint scientific consultations, identification of innovative health technologies, and voluntary cooperation beyond the scope of the Regulation.
Filling in the framework
The three years between the adoption of Regulation (EU) 2021/2282 and the first batch of JCAs were devoted to the voluntary EUnetHTA21 (European Network of Health Technology Assessment) consortium, along with member states and the European Commission, filling in the regulation’s legal and organisational framework with processes and methodologies for HTA harmonisation. EUnetHTA21 ceased operations in September 2023, to be succeeded by the HTA Coordination Group
What the HTA regulation does not allow for is a single, centralised HTA procedure that might resolve persistent disparities in assessments across the EU. JCAs are limited to assessing comparative clinical safety and effectiveness, although choices of comparator products and endpoints for assessment can be made at EU level. Decisions on cost-effectiveness, drug value or budget impact, along with associated pricing and reimbursement negotiations, remain the responsibility of individual member states.
Nor are JCAs legally binding on member states, even though industry participation in the procedure is mandatory. Members are only required to give due consideration to JCAs when conducting national HTA procedures. Understandably, these grey areas have made industry nervous.
Complications include differing national conceptions of the standards of care used as comparators for clinical effectiveness, or disagreement over the validity of surrogate endpoints applied in clinical trials. Although the regulation is expected to level up assessments in member states where HTA capacity and expertise is underdeveloped (e.g. in Eastern Europe), there are fears that it may lead to even more duplication of work or further complicate the market access landscape.
Plan ahead but stay agile
Deloitte’s advice to market access teams is to “plan ahead but stay agile”, with particular attention to pre-launch preparation, new modes of strategic decision-making, cross-functional collaboration and the involvement of local teams. While the HTA regulation will require end-to-end changes in asset development, affecting all cross-functional teams, the level of disruption will vary from country to country, depending on the current maturity and willingness to adapt to local HTA procedures, Deloitte says.
For planning purposes, it divides these countries into five archetypes: Safeguard countries (e.g. Germany, Denmark), Timesavers (France, Italy, Belgium), Discoverers (Spain, Portugal) and Backbenchers (Lithuania, Estonia, Greece), plus Observer countries outside the EU that may take into account published JCAs in their own assessments. In Timesaver and Discoverer countries, there should be more potential to accelerate HTA processes on the back of the HTA regulation, even if this implies increased disruption for local HTA submissions, Deloitte notes.
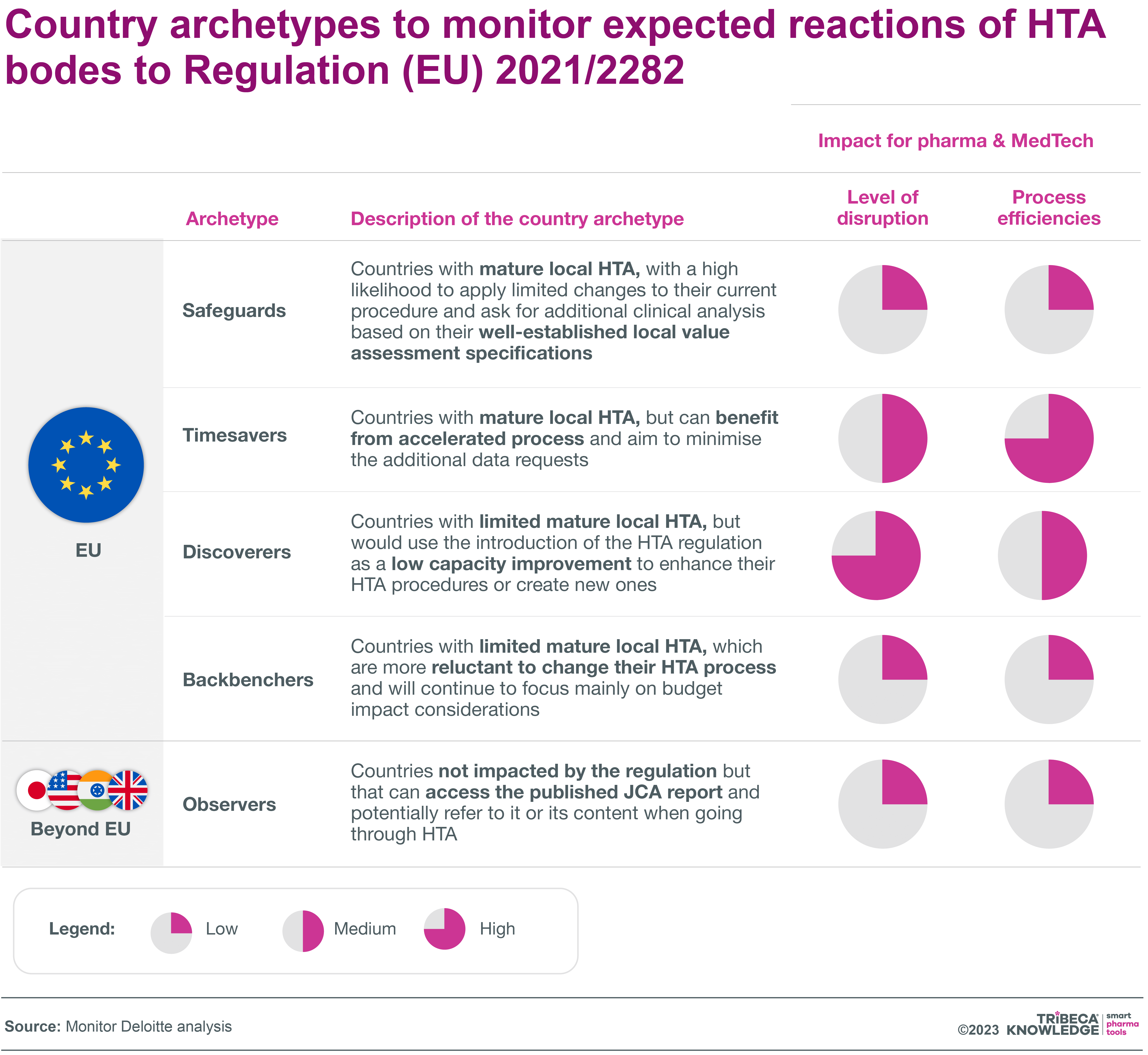
Source: Monitor Deloitte analysis
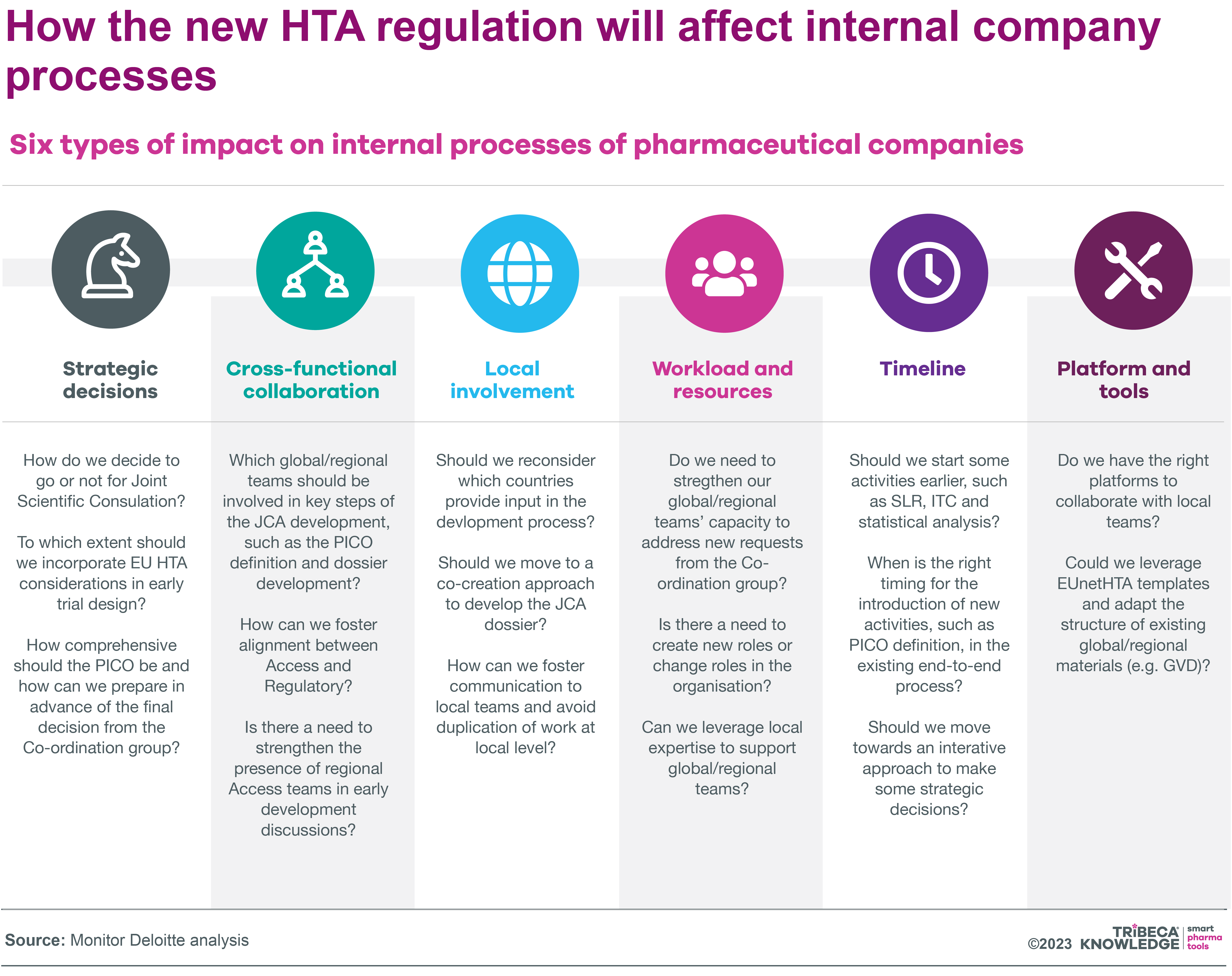
Deloitte’s report also looks at how the HTA regulation is likely to affect MA and HTA processes within companies, identifying six key areas of impact:
- Strategic decision-making at critical junctures in drug development: e.g. go/no-go decisions for joint scientific consultations, balancing HTA requirements versus regulatory requirements in determining early clinical-trial design)
- Cross-functional working: g. strengthening collaboration with global and regional cross-functional teams, and between market access and regulatory specialists, from early development through to preparation and submission of JCAs
- Local involvement: g. building a centralised process in which local needs are considered early on in crafting a comprehensive clinical-effectiveness approach to JCAs and local HTA dossiers
- Workload and resources: e.g. re-assessing time and workload commitments of global and regional teams, developing capacity-forecast capabilities, creating new roles to facilitate and co-ordinate JCA preparation and submission
- Timelines: e.g. accelerating existing pre-launch activities to accommodate JCA preparation and submission in parallel with the regulatory-approval process; aligning these with new activities like engagement with EU clinical experts.
- Platforms and tools: e.g rethinking existing technology and collaboration platforms to advance JCA submission and delivery, redefining global document structures based on EUnetHTA templates
Source: Monitor Deloitte Analysis
PICO = Population, Intervention, Comparison, Outcome (Standard format for defining research questions)
As Deloitte stresses, the time to act on these issues is now, even if uncertainties remain around final methodologies, guidance documents or timelines for centralised HTA processes. Companies looking to optimise their HTA operations and increase speed to market in Europe from 2025 onwards should take these recommendations to heart.
Timely corporate engagement with new channels for pan-EU health technology assessment, both externally and internally, is vital if companies want to guard against lowest-common-denominator assessments or poorly aligned processes. Otherwise, the EU regulation risks adding complexity without streamlining timescales, enhancing outcomes, or achieving meaningful and remunerative HTA harmonisation across the member states.
What lies ahead
This blog on pharmaceutical trends for 2024 has outlined some important considerations for market access managers and their teams as they prepare for another year of change and consolidation in the marketplace. As always, the devil is in the details, and pharmaceutical interests are hard at work trying to limit potential damage to their businesses and the wider cause of abundant, available and affordable drug innovation.
Nonetheless, even with healthcare systems recovering from the extraordinary circumstances of COVID-19, uncertainties in the timing, coherence, criteria, and outcomes of HTA, pricing, and reimbursement procedures will continue to mould market access and launch strategies for new medicines in the foreseeable future. That calls for advanced digital transformation, better-informed, more responsive market access strategies, and better aligned yet agile launch execution.
Launch readiness software like SmartLaunch™, or digital tools geared to market access, such as SmartAccess™, can help to manage shifting timelines more efficiently, while providing real-time visibility of HTA, reimbursement and launch status across multiple countries and corporate layers. Given the pressing challenges highlighted in this pharmaceutical industry analysis 2024, companies need to be all the more confident that their products can be launched and adopted in the marketplace with the maximum possible impact.
One way to do that is by optimising digital solutions, and specifically collaboration software, to maintain alignment right across the board in pharmaceutical product launch and market access planning and execution. A 360⸰, real-time view of progress and challenges from market to market will help to ensure 2024 is a year in which market access hurdles are not only fully assimilated and understood, but leveraged to deliver competitive advantage for companies and truly beneficial outcomes for health systems and patients.
