
 Andre Moa
Andre Moa
 28 Nov 2023
28 Nov 2023
 15 minute read
15 minute read

The drug-approval slump in major markets that clouded innovation prospects during 2022 seems to be over. In this blog, you can find out more about 2023 new drug approvals by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA), and how those are translating into game-changing and commercially auspicious launches to watch out for during 2024.
The drug approval slump in major markets that clouded innovation prospects during 2022 seems to be over. In this blog, you can find out more about key approvals by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) in 2023, and how those are translating into game-changing and commercially auspicious launches to watch out for during 2024.
As the last months of 2023 played out, the sense of recovery in the approvals space was particularly acute in the all-important US market, despite potentially discouraging trends such as more stringent requirements for accelerated approvals and the introduction of drug price negotiations under Medicare.
Centralised approvals of pharmaceutical products in the European Union looked set to end the year more in line with the 2022 trend, or perhaps slightly ahead. In Europe too, the current overhaul of pharmaceutical legislation is raising questions about market access and the sustainability of industry’s business models. That said, EU drug approvals were not quite as dramatically curtailed as in the US during 2022.
A wide range of factors, including agency resources, the rate and complexity of approval submissions, and spillovers from one year to the next, can influence basic data on approval volumes. Nonetheless, these data are one barometer of the pharmaceutical industry’s commercial health and R&D productivity, whatever other challenges companies may face in the broader marketplace.

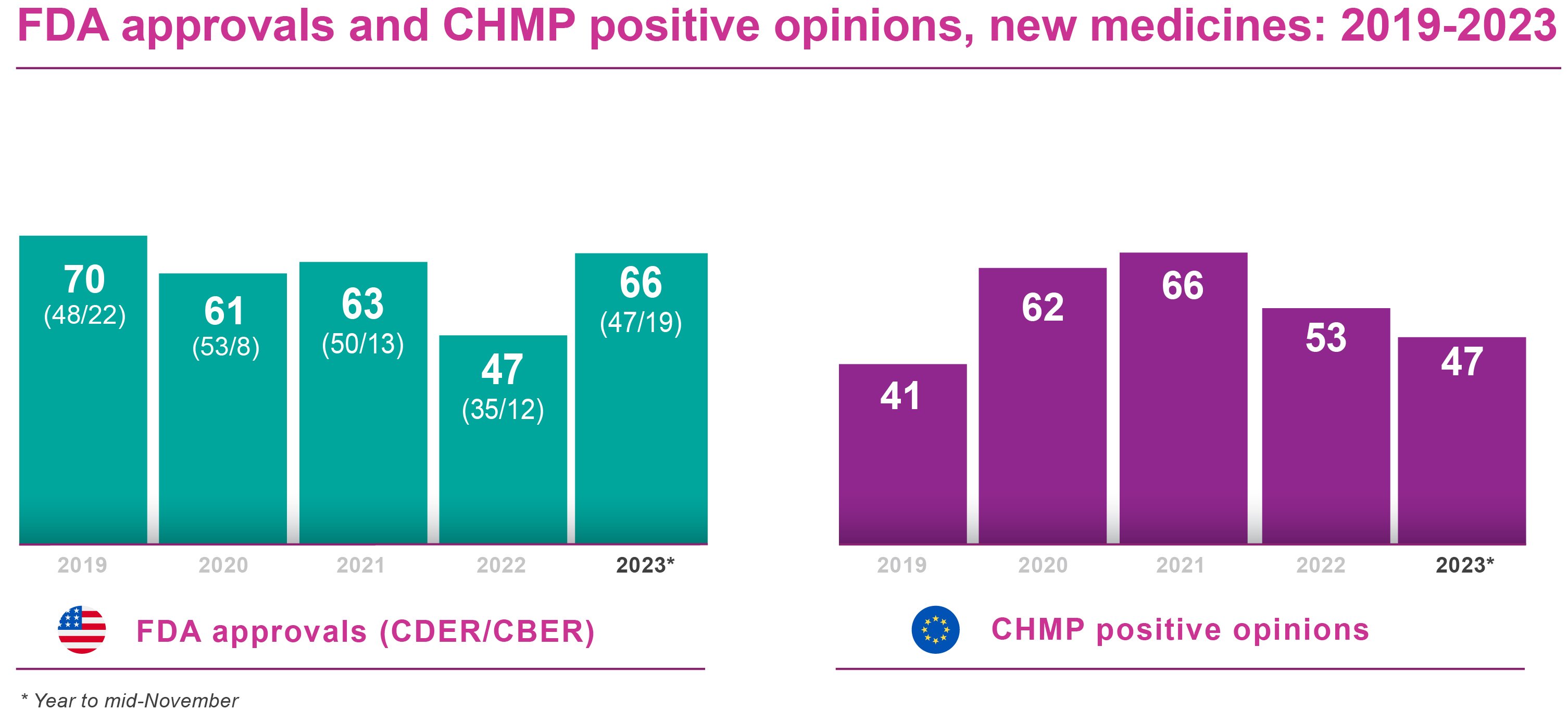
The FDA’s Center for Drug Evaluation and Research (CDER) had approved 51 new molecular entities and new therapeutic biologicals by the second half (H2) of November 2023. This was well ahead of the 29 products authorised by CDER as of mid-November 2022, and of the 35 approved by year-end. Moreover, it already surpassed the 47 drugs, vaccines and other biological products approved by CDER and the Center for Biologics Evaluation and Research (CBER) combined over the whole of 2022.
CBER reported 19 biological license application (including source plasma) approvals as of H2 November 2023. By comparison, CBER approved 10 products in the same period of 2022 and scored 12 approvals for the full year. The current year CBER approvals took the FDA’s year-to-date tally up to 70 products. The combined CDER/CBER count is already well ahead of 2021 and 2020, and should easily beat the 2019 full year tally of 70 FDA approvals overall.
On the European front, the EMA’s Committee for Medicinal Products for Human Use (CHMP) had recommended a total of 47 new medicines for approval by the second half of November 2023. There were 53 CHMP positive opinions in FY 2022, down from an exceptional 66 in 2021, and 49 by mid-November 2022. This suggested that, with one CHMP meeting still to come in 2023, the EMA might yet improve on its positive opinion count for 2022, albeit with a markedly less spectacular recovery than in FDA registrations.
In most cases, the European Commission concurs with the CHMP’s positive opinions. There may, though, be several months’ delay between CHMP recommendation and Commission sign-off. At the time of writing, 31 of the 47 new products recommended by the CHMP had secured marketing authorisations from the Commission, according to publicly available information.
Drivers for recovery in 2023
The reasons why major-market approvals fell so short in 2022 were not immediately apparent. Moreover, fluctuations in US drug approvals tend to draw more attention than year-to-year variations in the EU. Companies still treat the US as a testing ground for key assets, and as an opportunity to establish favourable price points and market-access strategies.
A study published last year by the Journal of the American Medical Association found that marketing authorisations for 95% of 89 new oncology therapies approved by both the FDA and EMA between 2010 and 2019 came in the US before the EU. Moreover, median review times for EMA approval of these therapies (426 days) were more than double the time taken (200 days) by the FDA.
The drivers for recovery in 2023 are also hard to pin down. The disruptive impact of the COVID-19 pandemic is now largely historical, although drug approvals held up remarkably well under pandemic conditions in 2021. On the other hand, it may be that the sharp focus on addressing COVID-19 needs during 2021 had a lingering impact on regulatory activity in 2022, as adjusted priorities, approval backlogs or staff shortages took their toll.
A brake on expedited approvals?
One theory about last year’s drought was that the FDA was treading more carefully on expedited approvals. The agency had sustained heavy criticism for clearing Biogen’s Alzheimer’s disease therapy Aduhelm (aducanumab) on the basis of surrogate endpoints in June 2022. Other accelerated US approvals have also proved contentious. In particular, there were claims of a growing tendency to approve new drugs based on single clinical trials, and with lower levels of public disclosure.
This perception gained some more credence in late December 2022. Legislation introduced as part of the Consolidated Appropriations Act, gave the FDA new authority to improve transparency around accelerated approvals and ensure that applicants complied with post-marketing requirements. Under the Food and Drug Omnibus Reform Act, for example, the FDA must specify conditions for post-approval studies by the date of the accelerated approval. The Act also creates a formal expedited withdrawal procedure for drugs approved via the accelerated approval pathway.
The FDA followed up in March 2023 with proposed guidance for cancer drug developers. This suggested that randomised controlled studies were “the most appropriate trial design” to support accelerated approvals of oncology therapies and could help accelerate confirmation of clinical benefit in post-marketing studies.
To some degree, the faster pace of US new drug approvals for 2023 allays fears that a tougher FDA stance on expedited submissions might chill medicines innovation. Nonetheless, there are indications that the less permissive stance is already having an impact in the US, and will continue to do so in years to come.
No such parallel initiatives have emerged to date in the European Union, where expedited regulatory pathways (ERPs) are used less frequently than in the US. In fact, the European Commission’s wide-ranging revision of pharmaceutical legislation includes proposals to reduce the time taken to evaluate new medicines centrally in the EU.
The EMA would have to assess these products within 180 days (150 days for medicines of ‘major public health interest’), down from 210 days at present. The European Commission would then have 46 rather than 67 days to give its seal of approval. There are also plans for regulatory ‘sandboxes’, with adaptive frameworks including real-world data collection, to expedite approvals of novel therapies.
Breakthrough innovation
Volumes apart, the approval lists for new medicines in 2023 suggest that breakthrough innovation continues to be a strong component of US and EU approvals. The lists again include cell or gene therapies, as well as other novel approaches to previously intractable or inadequately managed diseases, such as Alzheimer’s. Examples include:
- BioMarin Pharmaceutical’s Roctavian (valoctocogene roxaparvovec-rvo) a gene therapy for the treatment of severe haemophilia A in adults. The FDA approved Roctavian in June 2023, following the conditional marketing authorisation granted to BioMarin’s product by the European Commission in August 2022.
- Sarepta Therapeutics’ Elevidys (delandistrogene moxeparvovec-rokl), the first gene therapy for paediatric patients aged four to five years with Duchenne muscular dystrophy (DMD) and a confirmed mutation in the DMD gene. The FDA granted Elevidys accelerated approval in June 2023, a decision that attracted some controversy. A European submission is awaited.
- Eli Lilly’s Omvoh (mirikizumab-mrkz), the first interleukin-23p19 antagonist for the treatment of moderate to severe ulcerative colitis in adults. The FDA approved Omvoh in October 2023, after the European Commission cleared the drug for marketing in May 2023.
- Pfizer’s Abrysvo, the first respiratory syncytial-virus (RSV) vaccine for passive immunisation of infants from birth to six months, following administration to the mother during pregnancy. Abrysvo, which secured FDA approval for older adults in May 2023, and for maternal immunisation in August 2023, is also indicated for active immunisation of adults aged 60 years and over to prevent lower respiratory-tract disease caused by RSV. The European Commission cleared Abrysvo for both indications in August 2023.
- Eisai/Biogen’s Leqembi (lecanemab-irmb), an amyloid beta-directed antibody for the treatment of adult Alzheimer’s disease. The FDA granted Leqembi accelerated approval in January 2023, and subsequently traditional approval in July 2023. In Europe, a CHMP decision on Leqembi is pending.
The shift to specialty drugs
Many of the breakthrough medicines listed above are indicated for rare diseases, consolidating a trend in recent years for R&D to focus on premium-priced specialty therapies, and in particular on orphan drugs addressing unmet needs. Of the 70 new medicines approved by the FDA in the year to date, for example, 37 have been orphan-designated. In the whole of 2022, 37 or 54% of just CDER’s novel drug approvals were for products addressing rare diseases.
By mid-November 2023, the CHMP’s 47 approval recommendations in the European Union included 22 orphan-designated products. Of the 53 products recommended for approval by the CHMP in all of 2022, 25 were orphan medicines.
Recent trends in drug development and approvals, such as breakthrough therapies for Alzheimer’s disease and obesity, may suggest something of a swing back towards high-impact new medicines aimed at large patient populations. Some regulatory tendencies of particular interest to industry could prompt re-appraisals of R&D and commercial strategies.
These include:
- Mandatory negotiations on Maximum Fair Prices (MFPs) for high-cost drugs used in the US Medicare system
- Rebate penalties for above-inflation price increases on Medicare drugs
- Potential reductions in regulatory data protection periods under the EU’s pharmaceutical review, unless new medicines are launched in all member states within two years of marketing authorisation
- Potentially restrictive criteria for unmet or high unmet medical need in EU approvals that might qualify new drugs for additional RDP or orphan-market exclusivity
In the US, for example, rebates on above-inflation price increases in Medicare could tip the balance towards volume-driven strategies. Alternatively, the limited grace periods available before MFP negotiations take effect might dampen enthusiasm for current ‘pipeline-in-a-product’ strategies geared to cumulative ‘indication-stacking’ and progressively larger patient populations. That said, the MFP provisions do significantly favour biologics, the heartland of the specialty-drug category, over small-molecule drugs in the time available before high-cost products are subject to MFPs.
Potentially shorter RDP periods in the EU could also undermine indication-stacking for orphan drugs, even if the declared intention of the Commission’s review is to stimulate innovation and competitiveness. Companies may have to think about how best to recoup R&D investment within the more limited period before generic or biosimilar competition weighs in.
For all that, current indications are that the speciality drug model remains alive and kicking. According to pharmaceutical analytics and consulting firm IPD Analytics, nearly 80% of the new medicines expected to be approved by the FDA during 2023 are specialty drugs, and at average prices ranging from $200,000 to $400,00 per year.

Blockbuster potential
For companies with innovation to sell, pharmaceutical launch success inevitably comes down to revenues and profit margins. Clarivate’s Drugs to Watch report for 2023 lists 15 products either already launched or scheduled to launch in 2023, and likely to achieve blockbuster status (annual sales of US$1 billion or more) and/or transform treatment paradigms within five years.
That compares with the seven potential blockbusters included in Clarivate’s Drugs To Watch report for 2022. It should be noted, though, that Clarivate revised its methodology for 2023 to include not only potential blockbusters, in strictly commercial terms, but innovative drugs. According to current forecasts, these latter might fall short of $1 billion in sales over the next five years but have the potential to transform treatment paradigms.
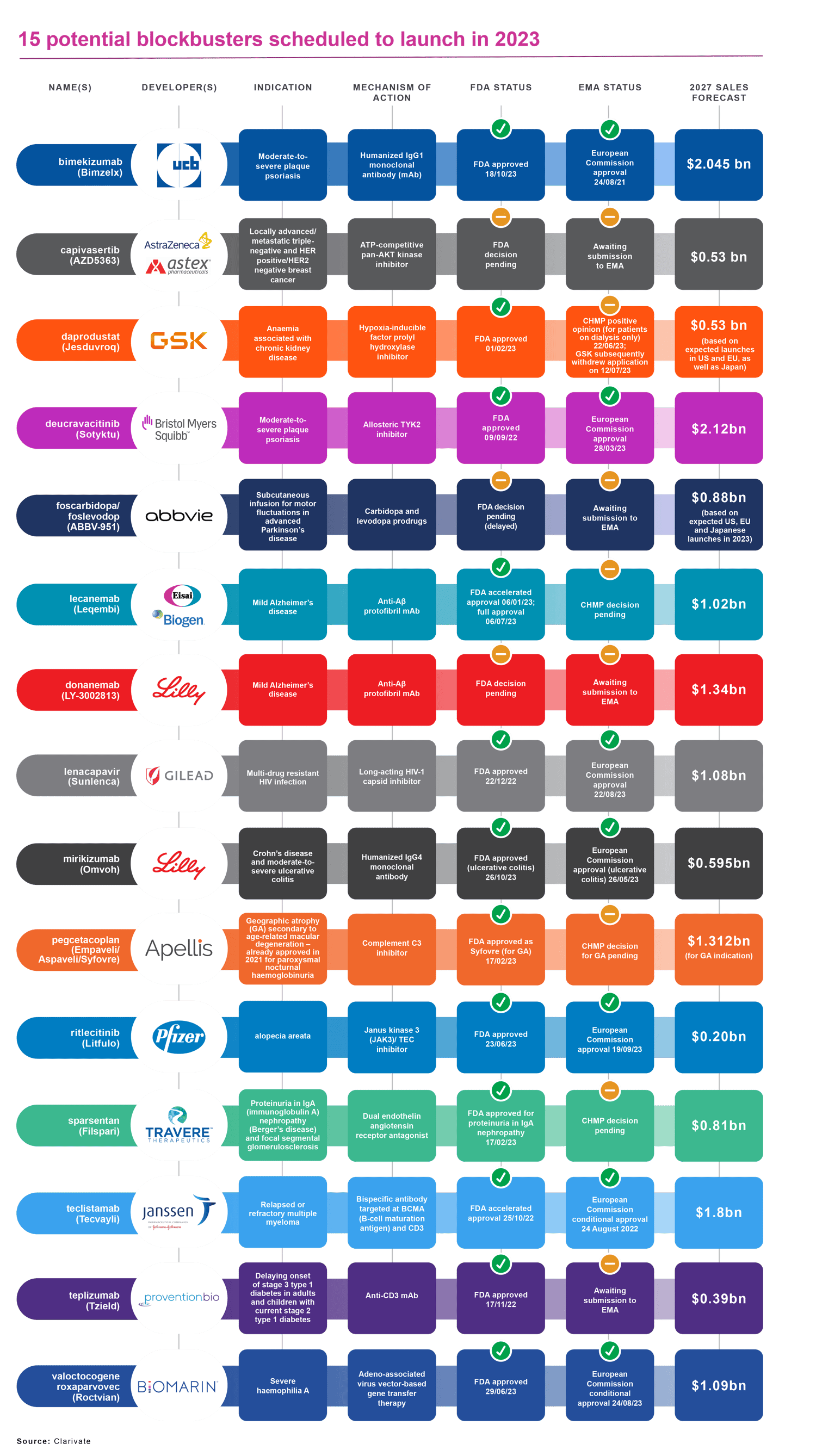
Source: Clairvate
From R&D to launch excellence
In the meantime, drug development pipelines continue to expand, albeit a little more slowly than in 2022. According to Citeline Clinical’s Pharma R&D Annual Review 2023, a total of 21,292 drug candidates were in active development as of early January 2023, 5.89% more than in the previous year’s analysis, when pipeline growth year on year was 8.22%.
As the report noted, though, the 2023 pipeline growth rate was an improvement on the 4.76% seen in 2021 and not far behind the five-year average growth rate of 6.90%. Moreover, the number of pipeline assets in Phase III was up by 9.8%, compared with an 8.7% Phase III gain in 2022 and 0.9% year-on-year growth in 2021.
All of this suggests that industry’s R&D engines continue to drive drug innovation with potential to transform patients’ lives. Yet many challenges remain in the gap between regulatory approval to market access. Companies must ensure on every front that R&D productivity translates into drug approvals, and approvals into successful launches.
This calls for the right launch management tools, such as SmartLaunch™, to ensure that milestones, timelines and challenges in pharmaceutical launch plans are visible, coordinated and actionable in real-time across different countries, functional silos and layers of management. Only with this kind of alignment can regulatory approvals set the stage for launch excellence. As major-market approvals fluctuate from year to year, the capability to make every launch count has never been more important.
