
 Andre Moa
Andre Moa
 8 Dec 2022
8 Dec 2022
 54 minute read
54 minute read

The pharmaceutical industry has little room for complacency in a post-COVID world. As we map out in our pharma trends for 2023, COVID-related disruption has created a whole new set of launch readiness and market access challenges for companies looking to maximise their assets in the new year. Moreover, it has changed some market characteristics for good. Patient journeys are permanently altered, while remote working is now firmly embedded in working practices, boosting the uptake of digital tools.
Moreover, it has changed some market characteristics for good. Patient journeys have permanently changed, and remote working is embedded, thus boosting the uptake of digital tools.
For much of the developed world, the pandemic looks close to exiting its acute phase, or at least in steady transition from pandemic to endemic status. All the same, governments and populations need to stay alert to the possibility of resurgence and further viral variants. Pharmaceutical companies would also be ill-advised to drop their guard.
In a healthcare environment absorbing the long-term impact of COVID management, the emphasis is now more than ever on cost controls and efficiency gains that will bear down on pharmaceutical market staples such as pricing and reimbursement. There are also strong incentives to shift healthcare strategy more decisively from treatment to prevention, contrary to the long-established pharma business model. A growing burden of age-related diseases has enormous cost implications, even as pharma debuts the first disease-modifying therapies for conditions such as Alzheimer’s.
The pandemic shook up healthcare systems and their suppliers on multiple levels, including barely sustainable pressures on healthcare capacity and finances, diversion of funds to pandemic-related needs, staff shortages, interrupted supplies, new and urgent R&D priorities, digital transformation and accelerated virtual interaction. That disruption has also planted the seeds for new ways of working and has delivered an innovation dividend that should help propel the best-prepared and the most agile, creative and open-minded pharmaceutical companies through the difficult years ahead.

Pharmaceutical Trends for 2023
Trend #1. Negotiating the post-COVID launch and market access environment
As we enter 2023 with perennial hopes of fresh starts and renewed energies, COVID-19 has inevitably left a deep footprint. There is much that is either new or augmented for the industry to negotiate in the product launch and market access landscape. As we consider the key trends shaping pharma in this uncertain climate, one residue of the pandemic has been to change the whole nature of the patient journey, from diagnosis through to medical intervention and pharmaceutical therapy.
The pathway to diagnosis and treatment has become “lengthier, leakier, and more complex”, note Sarah Rickwood and Kirstie Scott, respectively, IQVIA’s Vice President, EMEA Marketing and Thought Leadership, and Consultant, EMEA Thought Leadership, in a white paper on Overcoming Pharma’s Launch Performance Problem. Even oncology, the therapy area generating the highest number of innovative launches, has run up significant backlogs due to delayed or cancelled screening, diagnosis and treatment.
"The pathway to diagnosis and treatment has become “lengthier, leakier, and more complex”"
Moreover, a decline in face-to-face patient consultations and a marked (in many instances, permanent) shift to remote patient management has reduced opportunities to evaluate treatment progress and update therapies. Doctors are generally reluctant to prescribe newly launched medicines remotely, Rickwood and Scott observe.
Among other trends to watch, the pandemic has consolidated the impact of stricter institutional or regulatory constraints on pharmaceutical companies’ access to healthcare professionals. According to a recent McKinsey survey, the industry’s in-person interactions with doctors have fallen by 75% in Europe, while doctors expect two-thirds of their contacts with pharmaceutical sales representatives to continue occurring via virtual channels.
Furthermore, three out of four American healthcare professionals believe restrictions on rep visits are here to stay, McKinsey says. This could “hamper companies’ efforts to keep doctors informed about new drugs that may elevate the standard of care, which in turn would delay better outcomes for some patient populations”, warn McKinsey analysts Nils Peters, Pablo Salazar, Arnold Scaglione and Martin Uriarte.
Tougher launch environment
The analysts also point to the strong growth of launches into multi-indication categories (e.g. oncology, immunology) as something likely to test industry’s mettle in years to come. This presents additional challenges in areas such as co-positioning, pricing, sequencing of indication expansion, prioritisation of field-team resources, and orchestrating internal collaboration, the analysts note.
The evolving launch environment will be particularly tough on less experienced players, McKinsey believes. Its research suggests that first-time market entrants, which are expected to account for more than half of blockbuster launches between 2021 and 2025, have a significantly lower chance of success than their more experienced peers.
At the same time, even launch veterans recognise the need to diversify to stay ahead. Eighteen of the top 20 pharma companies have gene therapies in development, while 36% of launches, up from less than 5% in 2019, are expected to involve new therapeutic modalities by 2025. New territories, such as the fast-expanding Chinese market, are also an important part of the mix.
Reforms at the National Medical Products Administration boosted China’s annual approvals of innovative medicines from just seven in 2016 to 69 in 2021. With a growing population of patients in need, the Chinese pharmaceutical market is now second in size only to the US, even if much of the local market still comprises generics and off-patent drugs.
"Even launch veterans recognise the need to diversify to stay ahead. Eighteen of the top 20 pharma companies have gene therapies in development, while 36% of launches, up from less than 5% in 2019, are expected to involve new therapeutic modalities by 2025"
Inflation and cost-containment
Prominent among other launch and pharma market access challenges spawned by the pandemic, and likely to persist into 2023, is rampant inflation in many countries. This affects everything from R&D and manufacturing costs through to drug prices and access to medicines. Global inflation is estimated to have reached a record 12.1% in October 2022, according to Moody’s Analytics, albeit with some confidence that consumer prices would slow down from that point.
Nonetheless, inflation is expected to remain above target rates. With healthcare systems still working through post-COVID backlogs, drug budgets could come under even closer scrutiny, with growing demand for meaningful, demonstrable value from new medicines. In markets (including the US) where access to innovation may come with substantial co-payments and/or deductibles, patient face tough choices over how much of their disposable income they can spare for medicines when energy or food may make more urgent demands on personal finances.
As Rickwood and Scott point out, three of the largest single markets for prescription medicines – the US, Japan and Germany – are embracing further cost-containment measures. The US is already there, having signed the Inflation Reduction Act of 2022 into law last August (see Trend #2 below). In October 2022, the German Parliament (Bundestag) approved a draft act on the Financial Stabilisation of the Statutory Health Insurance System (GKV-FinStG), which could cost the pharmaceutical industry an estimated €4 billion.
"In Europe overall, reimbursement decisions on new medicines and indications increasingly come with conditions and restrictions attached"
This includes measures to limit free pricing for newly launched medicines to six months from the current 12; lower from €50 billion to €30 million the annual turnover threshold above which orphan drugs are subject to full benefit assessments in Germany; a four-year extension of the current reimbursement-price freeze; an increase from 7% to 12% of company sales in the mandatory discount to statutory health insurers; a mandatory 20% discount on new combination therapies; and more restrictive reimbursement prices for medicines without a significant added-benefit rating.
In Europe overall, reimbursement decisions on new medicines and indications increasingly come with conditions and restrictions attached, the IQVIA authors note. In Japan, meanwhile, additional “off-year” price revisions for pharmaceuticals, over and above the regular biennial price revisions, were implemented in 2021 and are likely to occur again in 2023.
Reshoring pharma manufacturing
One consequence of supply-chain interruptions during the pandemic has been to escalate concerns, in major markets such as the US and Europe, about over-reliance on countries like China (especially for active pharmaceutical ingredients/APIs) or India (APIs and generics) as sources for large swathes of the pharmaceutical market. A wide-ranging US Biden Administration review in June 2021 noted that, as of March that year, 52% of all finished dosage-form manufacturing facilities registered by the Food and Drug Administration (FDA) were outside the US, along with 73% percent of all FDA-registered API manufacturing facilities.
In October 2022, the European Commission released a working document on the Vulnerabilities of the global supply chains of medicines. “When trade disruptions or unanticipated demand surges threaten the supply of critical medicines and their raw materials or APIs,” it states, “the existence of sufficient EU manufacturing capacity can contribute to reducing supply vulnerabilities and ensuring supply security in the EU.”
The years to come may see growing efforts to mitigate supply-chain or perceived quality risks by ‘reshoring’ pharmaceutical production closer to the point of delivery. This is against the grain of historical outsourcing to low-cost manufacturing sites in developing countries.
While tax incentives and/or penalties may give some momentum to reshoring, these adjustments will also carry a substantial burden of time, costs and regulatory procedures. As with other critical components of market access, individual company strategies need to weigh carefully weigh the long-term gains of localisation against the investments needed to break the offshoring cycle.
"The years to come may see growing efforts to mitigate supply-chain or perceived quality risks by ‘reshoring’ pharmaceutical production closer to the point of delivery."
COVID-19 impact on launch performance
As the IQVIA white paper makes clear, the additional challenges posed by COVID-19 have further complicated the transition from regulatory approval to market access and launch excellence. In fact, approval rates for new active substances at both the FDA and the European Medicines Agency were historically high versus the pre-COVID average in 2020 and 2021 (a trend that did not, however, persist into 2022), while HTA decision-making in key European markets such as France, Germany and the UK more than kept pace during 2021.
"The additional challenges posed by COVID-19 have further complicated the transition from regulatory approval to market access and launch excellence"
In most markets, innovative medicines continued to launch during 2020-21 at volumes higher than, or very similar to, pre-pandemic benchmarks, Rickwood and Scott observe. Yet these launches underperformed substantially. Across eight countries that provide more than 90% of a new medicine’s commercial potential within the first five years of launch, average sales after six months were 19% below the pre-pandemic benchmark. Even if the traditional six-month performance test for launches may not be as relevant now as it once was, this figure suggests that current launch models require more than cursory adjustment.

Three pillars of post-pandemic launch excellence
How, then, can companies ensure that inflationary, budgetary, supply-chain or other post-COVID complications do not end up hampering access to genuinely valuable new medicines? For Rickwood and Scott, the only really effective way to navigate such a demanding and competitive launch environment is by combining speed of development and commercialisation with the three pillars of post-pandemic launch excellence set out in
Figure 1. 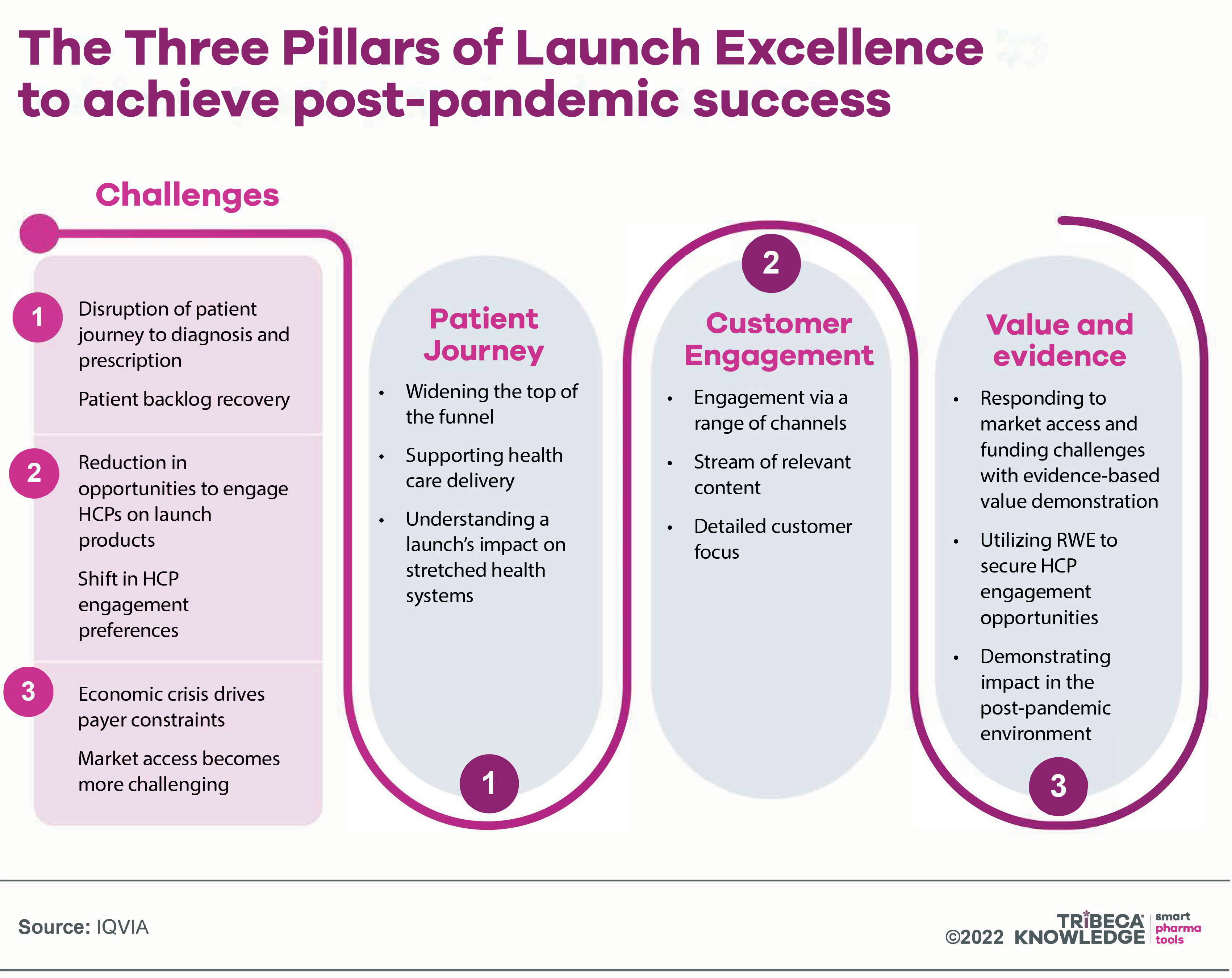
Source: IQVIA
That means first understanding in detail how the patient journey has altered in the shadow of COVID-19 and overcoming associated barriers to diagnosis and treatment. Disease-awareness campaigns, patient screening and better patient-support programmes may all help to ‘widen the top of the funnel’, by motivating healthcare professionals (HCPs) to identify patients and patients to seek care.
Limited healthcare-system capacity, aggravated by COVID-19, is another disincentive to treatment optimisation. Companies can ease this barrier through support for healthcare and medicines delivery (e.g. partnerships with homecare providers and speciality pharmacies), leveraging new technologies such as remote monitoring and health wearables. They must also be sensitive to a new launch’s potential impact on hard-pressed healthcare resources, offering innovative solutions such as population health-management strategies that target high-risk patients.
Companies then need to claw back some of the interactive engagement (both virtual and face-to-face) with HCPs which, in most key markets, has yet to recover to pre-pandemic levels. That is about ensuring and orchestrating engagement via a wider range of channels, providing a stream of real-world evidence (RWE) on drug value and relevance, and tailoring both content and messaging to individual HCP preferences and needs.
The third pillar of post-pandemic launch excellence is a strong integrated evidence strategy, again drawing on RWE. For example, with ever more stringent conditions for market access and medicines funding, RWE can help to identify proactively sub-populations where a drug is likely to be most effective and to deliver the best outcomes. It is particularly useful in informing HTA assessments and payment-by-results schemes, both increasingly familiar characteristics of the market access landscape.
"Early pandemic drug launches that have underperformed may now find it difficult to retrieve lost commercial opportunities"
According to Rickwood and Scott, early pandemic drug launches that have underperformed may now find it difficult to retrieve lost commercial opportunities. In future, they insist, companies must learn everything possible from launches that ride out pandemic-related difficulties, preparing early and meticulously for market entry across the three launch pillars described above. But there are no quick or easy wins, the authors warn: “The path to launch success will be extended; whilst companies must invest for the sprint, they must also prepare for a marathon.”
Trend #2. The Inflation Reduction Act: gearing up for US price negotiations
Challenges to the traditionally liberal pricing environment that has made the US such an attractive early launch market, in an age of increasing international price referencing, have been on the agenda for many years. Political resistance and a powerful pharmaceutical lobby have generally kept the more invasive of these measures at bay.
The Trump administration, for example, was adamant about taming pharma prices in the US and advanced a range of initiatives to tackle pharmaceutical cost inflation. These included quite radical proposals, such as the ‘most favored nation’ plan to apply external reference pricing to medicines administered under the Medicare scheme. At one time, Trump also advocated giving Medicare authority to negotiate lower prices for new medicines.
However, few of these measures came to anything, while the ‘most favored nation’ rule was blocked after legal action from the pharmaceutical industry and other stakeholders. In this light, the drug-pricing provisions in President Joe Biden’s Inflation Reduction Act (IRA) are a particularly bold departure, even if more far-reaching proposals were shelved in the face of objections not just from industry and Congressional Republicans but from within Biden’s own party.
Signed into law last August, the IRA establishes for the first time the principle of direct price negotiations in Medicare, which accounts for a substantial proportion of retail prescription drug spending in the US. Prescription drug expenditure via Medicare Part D is projected to reach $119 billion in 2023, accounting for almost one third of all US spending on medicines through the retail channel.
"The IRA establishes for the first time the principle of direct price negotiations in Medicare, which accounts for a substantial proportion of retail prescription drug spending in the US"
The US has long preferred market-oriented approaches to pharmaceutical cost-containment, such as horse-trading between pharmaceutical companies and health plans or pharmacy benefit managers over confidential discounts and rebates to secure preferential formulary placement. As Don Creighton, partner at management consultants Oliver Wyman, points out, that is “very different than engaging with a government payer that can leverage the full force of the Medicare population”.
The IRA provisions
Briefly put, the Inflation Reduction Act allows the US federal government to negotiate prices for selected high-cost drugs used in the Medicare scheme, mirroring provisions already available under health programmes run by the Departments of Defense and Veterans Affairs and the Indian Health Service. The Act also puts an annual ceiling of $2,000 on out-of-pocket spending for prescription drugs under Medicare. Drug manufacturers will have to pay a rebate to Medicare if they raise their drug prices at rates above inflation.
Moreover, a new Manufacturer Discount Program will apply to selected brand-name medicines, biologics and biosimilars under Medicare Part D, both in the initial coverage and the catastrophic phases of the scheme’s prescription drug benefit. This will replace the existing Coverage Gap Discount Program with effect from 2025, mandating discounts of 10% off the negotiated price in the initial-coverage phase and 20% in the catastrophic phase.
The IRA applies to medicines administered under both Medicare Parts B and D, although only qualifying products in Medicare Part D will be affected in the first two years of the programme. The initiative begins in earnest during 2023, when the Centers for Medicare and Medicaid Services (CMS) will identify the 100 most expensive drugs in Medicare and pick 10 for direct price negotiations (a list of these should be published by 1 September 2023).
The Health and Human Services Secretary will then negotiate ‘a maximum fair price’ (MFP) with the manufacturer of each selected drug. The initial negotiation period will run from 1 October 2023 to 1 August 2024, with the Department of Health and Human Services (HHS) publishing a definitive list of negotiated prices no later than 1 September 2024, around two months before the next US presidential election on 5 November 2024. MFPs for the first tranche of Medicare Part D products will come into play in 2026.
These MFPs will be maintained until an approved generic or biosimilar equivalent reaches the market. In 2024, the CMS will also select another 15 Medicare Part D drugs for negotiation on MFPs to take effect in 2027, and then 15 from Parts D and B in 2025, with negotiated prices applying from 2028. From 2029, the agency will choose another 20 Part D and Part B medicines annually for direct price negotiations under Medicare, with prices taking effect three years later.
Figure 2 -png.png)
Source: Inflation Reduction Act, L.E.K. analysis
Manufacturers failing to agree an MFP with HHS will be subject to a tax penalty, amounting to 65 to 95% of the prior year’s drug utilisation under Medicare. Manufacturers that do agree an MFP but fail to honour it will incur civil monetary penalties. Only single-source drugs or biologics may be selected for Medicare pricing negotiations, and only if they have been approved by the FDA for at least nine (small-molecule drugs) or 13 (biologics) years. Some single-source drugs or biologics are exempt from the programme, such as new formulations of a qualifying drug or therapies for just one rare disease.
IRA impact on pharma
The IRA drug programme is expected to save the US government anything between $100 billion and $456 billion over a decade. While that is, on the surface at least, good news for the US health system, pharmaceutical manufacturers are more than a little uneasy. Major players such as Eli Lilly, Merck & Co, Bristol-Myers Squibb and Amgen have already warned of material impacts on their revue and income. Lilly, for example, says the new provisions factored into its decision to discontinue the development of a small-molecule blood-cancer drug, LOXO-338.
"Major players such as Eli Lilly, Merck & Co, Bristol-Myers Squibb and Amgen have already warned of material impacts on their revue and income"
That is despite Congressional Budget Office estimates that the new provisions will prevent only 15 of 1,300 new drugs from reaching the US market over the next 30 years. There may be an element of grandstanding in claims that drug-price controls will seriously hurt pharmaceutical R&D. Moreover, companies cannot be too shocked by measures that have attracted widespread cross-party support among the American public.
Figure 3 -png.png)
Source: L.E.K. analysis, Biotechnology Innovation Organization, Informa (now Citeline) Pharma Intelligence, Quantitative Life Sciences
All the same, the IRA is a turning point in a market that typically provides a safe haven where companies can offset more stringent price controls in other countries.
One provision viewed as particularly counterproductive is the significantly shorter grace period awarded to small molecule drugs (nine years) versus biologics (13 years), before they fall subject to Medicare price negotiations. As a point of comparison, the average market-exclusivity period for a small-molecule drug is estimated at 14.4 years (20.5 years for biologics).
It may be a little disingenuous for the industry to claim that the IRA disincentivises small-molecule development, when the market has tilted decisively towards large-molecule biologics in recent years. Nonetheless, small-molecule drugs are generally less expensive to make, easier to administer (i.e. orally) and lower-priced. All of these factors could have perverse cost-inflation implications for Medicare. For larger companies, furthermore, a balanced, diversified product portfolio is likely to offer greater flexibility and leverage than one focused solely on biologics.
Reversing the development and launch model
Another consequence of the new provisions may be to reverse the development and launch model whereby, particularly in high-interest categories such as oncology, companies tend to introduce new molecules for small populations and as later lines of therapy. Subsequently, positioning expands into new indications, larger patient populations and earlier lines of therapy as the evidence base builds up.
This is likely to prove less attractive if the clock starts ticking for price negotiations after the first US approval, and with only a small patient base from which to recoup investment. Companies may then choose to shift away from this “pipeline-in-a-product” strategy towards prioritising multiple assets with the same mechanism of action, each targeting a different indication.
There are also a number of details still to be ironed out, with some further guidance expected in the first quarter of 2023. One key issue will be whether and how HHS, in negotiating MFPs for Medicare, will be conducting something akin to health technology assessment (HTA).
It will certainly be looking at the clinical effectiveness and costs of selected medicines. Whether that extends to cost-effectiveness, a broader consideration of health benefits and patient outcomes, or any added value, is currently unclear. That said, the IRA explicitly precludes recourse to the quality-adjusted life years (QALYs) methodology used in many HTA procedures across Europe and elsewhere.
"Until some of the fine print of the IRA provisions is clarified, the industry must at least ensure that its current R&D pipelines are geared to optimal launch effectiveness and tell a value story at every opportunity"
With more than three years still to go before the first wave of directly negotiated Medicare prices kicks in, the pharmaceutical industry has enough time to strategise on gaming the new system or, potentially, challenging it through the courts. While some market observers believe the IRA drug-pricing programme will, at least initially, put only a small dent in the industry’s operations, the long-term implications for the US market and pharmaceutical business models should not be underestimated.
According to management consultants ZS, the “risks to drugmakers are significantly greater than many appreciate”. With the IRA’s impact growing over time and hitting US-focused operations in particular, the new provisions could even nudge companies into mergers and acquisitions, Moody’s analysts suggest. For the moment, and until some of the fine print of the IRA provisions is clarified, the industry must at least ensure that its current R&D pipelines are geared to optimal launch effectiveness and tell a value story at every opportunity.
Trend #3: Will 2023 see HTA harmonisation installed as the new reality for pharma market access?
Value is always subjective. The complexities of identifying, characterising, measuring and rewarding value are all the more challenging when applied to such a personal and emotive, yet politically and economically consequential, field as health and medicine.
Value can mean different things to pharmaceutical companies, regulators, healthcare professionals (HCPs), payers, patients, politicians or society at large. Even a nominally patient-oriented value such as quality of life may incorporate a vast range of subjective perceptions and preferences, depending on variables such as age, life circumstances and the particularities of a disease from one stage to the next.
For their part, pharmaceutical companies want to see a more holistic understanding of drug innovation, as set out in ISPOR’s ‘Value Flower’. This would extend beyond the relatively narrow value judgments currently applied to medicines by HTA agencies. Ideally, it would recognise the full range of long-term cost offsets and societal gains available from truly game-changing products.

Small wonder, then, that the heterogeneity of health technology assessment (HTA) procedures has complicated launch uptake and impact as value assessment of medicines proliferates worldwide. These mechanisms look at different elements of a drug’s value proposition (e.g. unmet need, clinical effectiveness, degree of innovation, budget impact, cost versus benefits, offsets to health systems and society). At the same time, there is no standardised global definition of value for medicines.
Continuing disparities inevitably make for inconsistencies and delays in market access across national borders, muddying launch planning and the development of broadly applicable value propositions for new medicines. According to the European Federation of Pharmaceutical Industries and Associations (EFPIA), for example, divergence in the time it takes patients to access new medicines in different EU member states is only getting worse.
The association’s latest and most comprehensive EFPIA WAIT Indicator Survey found that the average time between marketing authorisation and availability, or inclusion of a centrally approved medicine on a member state’s public reimbursement list, was 511 days as of 1 January 2022, versus 504 days in 2020. Patient access inequities ranged from around 133 days on average from approval to reimbursement in Germany, to more than 899 days in Romania.
An analysis of The root cause of unavailability and delay to innovative medicines, conducted for EFPIA by Charles River Associates (CRA), blames several factors, including regulatory process, pricing and reimbursement mechanisms, health-system readiness (e.g. available budgets), multiple layers of decision-making between national and regional approvals, and value-assessment or HTA procedures. Into this last category fall barriers such as misalignment on evidence requirements, misalignment between perceived or potential value and price (factoring in ability to pay), and differences in the value assigned to product differentiation and choice.
"Patient access inequities ranged from around 133 days on average from approval to reimbursement in Germany, to more than 899 days in Romania."
For example, the CRA/EFPIA report notes, misalignment on evidence ̶ “one of the most prominent and complex delaying factors” – can occur not only between industry, regulators and HTA bodies, but also between regulators and HTA bodies, or among different HTA bodies. It applies to the whole range of value assessment criteria, including patient populations, comparators, trial designs, endpoints, and statistical analysis. “Even once there is agreement on evidence, there can be a significant debate on whether this justifies the price of the medicine,” the report adds.
Figure 4 -png.png)
Source: EFPIA/Charles River Associates
The EU HTA Regulation
One light on the horizon in this respect is the European Union’s Regulation (EU) 2021/2282 on health technology assessment (HTAR), which came into force on 11 January 2022. The regulation does not actually apply until 12 January 2025. It creates a legal and organisational framework for co-operation between member states on EU-level joint clinical assessments (JCAs) of new or existing health technologies, including medicinal products, medical devices and in vitro diagnostics. Over the next three years, the EUnetHTA21 consortium, the member states and the European Commission will fill out this framework with processes and methodologies for more harmonised EU-wide HTA.
Overseeing the new JCAs will be a centralised Coordination Group with representatives from national HTA authorities. This replaces the voluntary European Network of Health Technology Assessment (EUnetHTA), which sought agreement between member-state agencies on common frameworks, principles and methodologies for HTA. The Coordination Group will also tap into a stakeholder network to include patient, HCP and industry associations, clinical and scientific societies, consumer organisations, and other non-governmental healthcare bodies.
Other than introducing JCAs, the HTA Regulation establishes a framework for joint scientific consultations (advice on evidence requirements for HTA submission), identification of innovative health technologies, and voluntary co-operation beyond the scope of the Regulation. The new rules will apply to all products approved centrally through the European Medicines Agency, as well as new indications of medicines subject to EU HTA assessments.
The JCA provisions start with novel cancer drugs and advanced therapy medicinal products (ATMPs) in January 2025, expanding to orphan drugs from 13 January 2028. All other medicines approved centrally through the EMA will be subject to JCAs from 13 January 2030. The procedure will involve companies submitting JCA dossiers 45 days before the Committee for Medicinal Products for Human Use (CHMP) issues a positive or negative opinion on EU-wide marketing authorisation.
The JCA report will then be published at the latest 30 days after the European Commission formally approves a medicine. As Alexander Natz, lawyer and secretary general of the European Confederation of Pharmaceutical Entrepreneurs (EUCOPE), points out, this suggests the EU’s JCA report could be among the first such clinical assessments for a new medicine globally, with potential for significant global impact.
Limitations of JCAs
That said, JCAs are still a long way from single, harmonised HTA procedures that will resolve the persistent disparities highlighted by EFPIA and other industry stakeholders. For one thing, they are limited to assessing comparative clinical safety and effectiveness. Decisions on cost-effectiveness, drug value or budget impact, along with associated pricing and reimbursement negotiations, remain the province of the member states.
"JCAs are still a long way from single, harmonised HTA procedures that will resolve the persistent disparities highlighted by EFPIA and other industry stakeholders"
That reflects varying national demographics, health profiles, budgetary resources, healthcare strategies and political priorities across the EU. On the other hand, Natz observes, choices on comparator products and endpoints for assessment will be made at EU level, which could have a significant influence on national pricing and reimbursement negotiations.
JCAs will also reduce the administrative burden on product developers through a single, one-time dossier for EU-wide clinical assessment. One disappointment for the industry, though, has been that JCAs are not legally binding on member states, even though industry participation in the procedure is mandatory. Members are only required to give due consideration to JCAs when conducting national HTA procedures.
Moreover, they can ask manufacturers for new data and evidence on the relevant product (even for new comparators), although not for any information already submitted as part of the joint assessment. Natz is confident, nonetheless, that “the acceptance of JCAs at national level will be high, as the national HTA bodies will be involved in the EU decision-making”.
If member-state HTA agencies do make additional requests for evidence after reviewing the JCA, this could delay both national HTA procedures and final reimbursement decisions, notes Chloe Sheppard, associate consultant at Partners4Access. The risk is likely to be less acute in countries without formalised HTA mechanisms, such as Greece, where national authorities will not have to take so much account of established procedures.
And while the EU regulation may feasibly encourage national bodies to refocus their HTA efforts on economic evaluations, that will require member states first to be fully aligned with the EU’s JCA methodology, Sheppard adds. In that respect, “is it possible to develop a clinical-assessment methodology which aligns with the perspectives of all member states who have historically had differing ideas about value and the acceptability of evidence?”, she asks.
Otherwise, national bodies may be inclined to ask for additional evidence in cases “where the standard-of-care treatment for a particular condition varies between EU countries, or a clinical-trial endpoint is accepted by one national HTA agency but rejected by another”, Sheppard comments.
Going global with HTA harmonisation
In short, pharmaceutical companies looking to launch products across the European Union may not be hoping for wholesale HTA integration, but will at least have a viable platform for further HTA harmonisation. As with the new Medicare pricing provisions in the US Inflation Reduction Act (see Trend #2 above), much remains to be ironed out, and the next few years will be critical in putting meat on the bones of the EU’s HTA regulation.
"Pharmaceutical companies looking to launch products across the European Union may not be hoping for wholesale HTA integration, but will at least have a viable platform for further HTA harmonisation"
In the meantime, some EU member states are already conducting joint HTA assessments of a kind, albeit generally limited to exceptional cases such as orphan drugs or other medicines expected to weigh heavily on drug budgets. One example is the BeNeLuxA collaboration between Belgium, the Netherlands, Luxembourg, Austria, Ireland and the Nordic Pharmaceutical Forum, which collaborates on horizon-scanning for new medicines, information exchange and health technology assessments, as well as pricing and reimbursement.
This has led to joint agreements on pricing, and subsequent pay-for-performance contracts at national level, for high-cost innovative therapies such as Zolgensma (onasemnogene abeparvovec), AveXis/Novartis’ gene therapy for spinal muscular atrophy (SMA); and Zynteglo (betibeglogene autotemcel), bluebird bio’s gene therapy for the treatment of severe beta thalassemia. There are also some recent indications of more international convergence on HTA.
Global market access consultant Neil Grubert cites, for example, a recent cross-border HTA alliance, the AUS-CAN-UK Collaboration Arrangement. This involves England’s National Institute for Health and Care Excellence (NICE); Healthcare Improvement Scotland (which oversees the Scottish Medicines Consortium); Health Technology Wales; the All Wales Therapeutics & Toxicology Centre; the Canadian Agency for Drugs and Technologies in Health (CADTH); and the Australian Government Department of Health and Aged Care (which oversees the Pharmaceutical Benefits Advisory Committee).
The collaboration includes, under the heading of work-sharing and efficiency gains, exploring the feasibility of recognising or using partner HTA information and running a pilot for joint clinical assessment. As Grubert comments, the alliance’s “work on mutual recognition of information and the pilot JCA will be followed with particular interest—not least at a time when the EU is expending enormous effort on preparing for a JCA system that has encountered considerable resistance”.
And what of the always important US market? The non-affiliated Institute for Clinical and Economic Review uses Quality-Adjusted Life Years, a standard cost-effectiveness measurement applied by NICE and many other HTA bodies, in its reviews of healthcare interventions. These assessments may exert some influence on payer-decision making and drug uptake. However, the most potentially disruptive threat to US pricing and coverage in recent years – maximum fair prices (MFPs) for selected medicines used in the Medicare scheme ̶ rules out using QALYs in negotiations with manufacturers.
So far, it remains to be seen whether the US Department of Health and Human Services (HHS) will consider cost-effectiveness or other typical HTA parameters in negotiating and setting MFPs. If it does, though, HHS might find it hard to ignore what is happening in the EU and other nerve centres of HTA proliferation and harmonisation.
Trend # 4: Real progress on diseases of ageing?
As populations age worldwide, the growing demographic mismatch between supply and demand presents a huge challenge to healthcare systems and funding, not to mention social-care provision, family dynamics and political decision-making. This is strongly evident in the US, for example, where, by 2035, 78.0 million of the population are expected to be aged 65 years and over, versus 76.7 million under 18 years old.
Countries such as India, Japan or China face an even heavier age-related healthcare burden. There were an estimated 414 million Asians aged 65 years and over in 2020, a figure projected to reach more than 1.2 billion in 2060. On a global scale, the proportion of the world's population over 60 years of age will nearly double from 12% to 22% between 2015 and 2050, according to the World Health Organization.
"As populations age worldwide, the growing demographic mismatch between supply and demand presents a huge challenge to healthcare systems and funding"
The cost of caring for older people rises sharply with age. In the US, for example, a recent health economic analysis calculated that average annual healthcare costs per capita swell from $2,062 during the first year of life to $14,307 in people still living at 85 years or over.
A key component of healthcare-cost inflation is age-related diseases with still limited treatment options, in particular debilitating and resource-intensive neurological conditions such as dementia, Alzheimer’s and Parkinson’s disease. Currently, more than 32 million people worldwide are estimated to be living with dementia due to Alzheimer’s. By 2030, this population is predicted to reach 78 million, at a cost to healthcare systems of $2.8 trillion.
Before even considering medical intervention, there are significant problems in initiating and mapping care pathways for Alzheimer’s and Parkinson’s patients, such as delayed and/or uncertain diagnoses. These challenges are aggravated by disease complexity, a shortage of reliable biomarkers, and reluctance on the part of healthcare professionals to deliver what is too often interpreted as a death sentence.
Currently, more than 32 million people worldwide are estimated to be living with dementia due to Alzheimer’s. By 2030, this population is predicted to reach 78 million, at a cost to healthcare systems of $2.8 trillion.
Once a diagnosis is made, though, the available treatment options for Alzheimer’s and Parkinson’s have still advanced little beyond creative variations on symptomatic relief. In the Parkinson’s arena, the last couple of years have seen disappointing clinical-trial results with two candidate monoclonal-antibody (mAb) therapies for disease modification.
Biogen’s cinpanemab, a mAb targeting the alpha-synuclein pathway, was discontinued in 2020 after failing to achieve its primary or secondary endpoints in the Phase II SPARK study. More recently, another promising alpha-synuclein inhibitor, Roche’s mAb prasinezumab, delivered disappointing results in the Phase II PASEDENA trial for early-stage Parkinson’s, although the compound is continuing in Phase IIb development.
Optimism for Alzheimer’s therapy
As things stand, the outlook for Alzheimer’s therapy is a little more auspicious. In particular, there has been a surge of excitement – and an ensuing backwash of scepticism – following the publication of full results from Clarity AD, a Phase III confirmatory trial with Eisai and Biogen’s anti-amyloid mAb, lecanemab, in early Alzheimer’s disease.
The Food and Drug Administration subsequently cleared lecanemab for US marketing as Leqembi through its accelerated approval pathway on 6 January 2023. Leqembi is indicated for patients with mild cognitive impairment or mild dementia due to Alzheimer’s. Eisai and Biogen filed for lecanemab approval as an early Alzheimer’s treatment with the European Medicines Agency on 11 January 2023.
Although lecanemab showed only modest benefits overall in Clarity AD, the outcomes were hailed as the first time a drug had both slowed the development of Alzheimer’s in the brain and reduced cognitive decline. It was also seen as a vindication, after years of setbacks, of the ‘amyloid hypothesis’ (see Figure 5 below), whereby abnormal accumulation of the protein beta-amyloid in the brain is seen as a principal cause of the disease.
Figure 5
-png.png)
Source: BBC Research
Specifically, the Clarity AD investigators reported that lecanemab met its primary endpoint of reducing clinical decline on the global cognitive and functional scale, CDR-SB. (Clinical Dementia Rating–Sum of Boxes). Mean changes from baseline in CDR-SB scores at 18 months were 1.21 on lecanemab and 1.66 on placebo, representing a 27% improvement with drug intervention.
Lecanemab also achieved statistically significant improvements versus placebo on its key secondary endpoints. In an amyloid PET sub-study involving 698 trial participants, for example, the mean change in centiloids (a scale for measuring amyloid imaging data) was -55.5 at 18 months for lecanemab and 3.6 for placebo.
Some scientists believe the effect size shown with lecanemab is still not enough to justify its risks. Alternatively, they want to see what happens with longer-term use of lecanemab, or caution that amyloids are only part of the picture in Alzheimer’s disease.
The road to treatment uptake
Eisai, which is taking the lead on clinical development and regulatory submissions for lecanemab, has been discussing the Clarity-AD data with regulatory authorities in the US, Japan and Europe. The aim was to file for traditional (i.e. non-accelerated) approval in the US and for marketing authorisation in both Japan and Europe by the end of March 2023.
At the least, the Clarity AD outcomes provide some much-needed hope for Alzheimer’s patients and a reprieve for Eisai and Biogen from the controversy surrounding the FDA’s accelerated approval of the companies’ amyloid-targeting predecessor Aduhelm (aducanumab) in June 2022. The European Medicines Agency turned down Aduhelm as an early Alzheimer’s treatment and Biogen withdrew its application for EU-wide approval in April 2022.
In the US market, the drug has struggled to achieve uptake and insurance coverage, despite a 50% reduction in Aduhelm’s price from $56,000 per year on launch. A further blow was the decision by the US Centers for Medicare and Medicaid Services (CMS) in April 2022 to reimburse the drug only for patients enrolled in clinical trials with Aduhelm.
This policy also has implications for other amyloid-targeting Alzheimer’s therapies approved on the basis of surrogate endpoints that predict but do not actually demonstrate clinical benefit. A more permissive approach would apply, though, to amyloid-targeting therapies (such as, potentially, lecanemab) that secure approval by showing direct clinical benefit in Alzheimer’s.
Even with FDA approval in the bag for lecanemab, effectively acknowledging evidence of the drug’s clinical benefit in modifying the Alzheimer’s disease pathway, we can still expect some difficult discussions about reimbursement provisions and other elements of market access. Particularly if lecanemab is premium-priced for unmet need, there could be debates over the ethics of distributing healthcare funds between competing diseases with high levels of age-driven demand (e.g. cancer).
In the US market, Leqembi has debuted at a list price of $26,500 per year. This is slightly below the revised average annual price for Aduhelm, but higher than the $8,500 to $20,600 annual price range estimated as cost-effective by the Institute for Clinical and Economic Review in a recent draft report. The CMS has indicated that it will consider covering Leqembi for Medicare patients, including those not enrolled in clinical trials with the drug, although the agency may wait for Leqembi to secure full-blown FDA approval first. Even then, CMS coverage could still be subject to evidence-development requirements.
"If the companies can show that early drug intervention with lecanemab actually does reduce the care burden and improve patients’ quality of life, with compelling measures of clinical and pharmacoeconomic benefit, then we really can start talking about a new era for Alzheimer’s disease."
The current care burden for Alzheimer’s is substantial and will only get heavier over time. Given their negative experience with Aduhelm, Eisai and Biogen will doubtless have thought carefully about launch planning for lecanemab and the hazardous road to treatment uptake. If the companies can show that early drug intervention with lecanemab actually does reduce the care burden and improve patients’ quality of life, with compelling measures of clinical and pharmacoeconomic benefit, then we really can start talking about a new era for Alzheimer’s disease.
Trend #5: Does pharma need to get more involved in disease prevention?
The pharmaceutical industry is built, for better or worse, largely on a well-honed model of incremental innovation. Research and drug development cycles are typically geared toward safer, more effective, better targeted or more patient-friendly treatments for conditions already addressed by other medicines.
There are exceptions, of course, such as a growing emphasis on treatments for rare diseases where there are no other options. Advanced therapy medicinal products (ATMPs), such as cell and gene therapies, have also broken the mould, offering the first hope of truly curative treatments that may require little more than a single (albeit challenging) medical procedure.
However, ATMPs still have several hurdles to cross, such as inherent complexity, the specialist expertise needed to administer treatments, and a relatively thin evidence base at present to dispel concerns about long-term efficacy. Moreover, cell and gene therapies tend to involve very high upfront costs that have to be mitigated by innovative payment systems
With healthcare costs still spiralling worldwide, and with further pressure from the legacy effects of the COVID-19 pandemic, disease prevention will become increasingly important, if not imperative, for healthcare systems to ensure sustainability. While the pharmaceutical industry has made various gestures towards a more holistic, integrated approach to patient care, such as disease-awareness campaigns or nursing support programmes, these efforts are usually linked in some way to driving treatment uptake.
"With healthcare costs still spiralling worldwide, and with further pressure from the legacy effects of the COVID-19 pandemic, disease prevention will become increasingly important, if not imperative, for healthcare systems to ensure sustainability."
The same applies essentially to companion diagnostic or biomarker development. They may encourage earlier and more precise identification and characterisation of disease, but in the interests ultimately of timely drug intervention. Arguably, this is in itself a preventive strategy, and a potential win-win for patients, health systems and pharmaceutical companies. Nonetheless, it does not really represent a wholesale commitment to disease prevention among the prevailing pharma market trends.
Vaccines, nutraceuticals, longevity medicine
The big exception is vaccines, which have more than proved their worth in steering COVID-19 from pandemic towards endemic status. There are also programmes underway to repurpose COVID-vaccine technology and develop mRNA vaccines for cancer. While vaccines are undoubtedly a source of substantial and more predictable revenue streams for industry, they carry their own challenges and remain a fairly specialised area of the pharmaceutical business.
Some pharmaceutical companies have also carved out niches in nutritional supplementation, functional foods and ‘nutraceuticals’, although again this is not generally a strategic priority for the industry. A more recent field that challenges the established pharmaceutical business model is longevity medicine. This is about using tools such as artificial intelligence, deep learning or biomarkers of ageing to develop individualised health plans and interventions, aimed at staving off age-related and chronic conditions.
There has been recent interest in widening public screening programmes for life-threatening diseases, in an effort to pick up those conditions early, when treatment is likely to have the most impact. The European Commission, for example, is updating its recommendations on cancer screening. This will include expanding population-based screening to lung, prostate and gastric cancers. In the UK, the Our Future Health study is collecting health and genetic data from five million adults for a long-term information repository to drive earlier identification, diagnosis and prevention of disease.
Disease prevention as a strategic imperative
Against this backdrop, could and should the pharmaceutical industry address disease prevention more as more of strategic imperative among its key market access trends in 2023? Theoretically, the industry could leverage its R&D expertise and massive data assets to position itself more explicitly as a solutions provider ̶ rather than just a negotiable cost of care ̶ along the whole healthcare continuum.
Alternatively, are prevention and well-being stretching the industry’s responsibilities a little too far? A concerted shift towards prevention would help to quiet accusations of disease-mongering. But would it not also risk killing the golden goose of incremental therapeutic, which for so long has defined and sustained pharma and its relationships with healthcare systems?
Indeed, there are indications that pharma is taking prevention seriously. When the Deloitte Center for Health Solutions asked biopharmaceutical executives in 2020 which of five emerging trends were likely to have the biggest impact on the life sciences industry over the next 10 years, non-pharmaceutical intervention came out as the leading threat to industry sustainability along with customised treatments. Prevention and early detection were seen as the next biggest threat (see Figure 6 below).
Figure 6
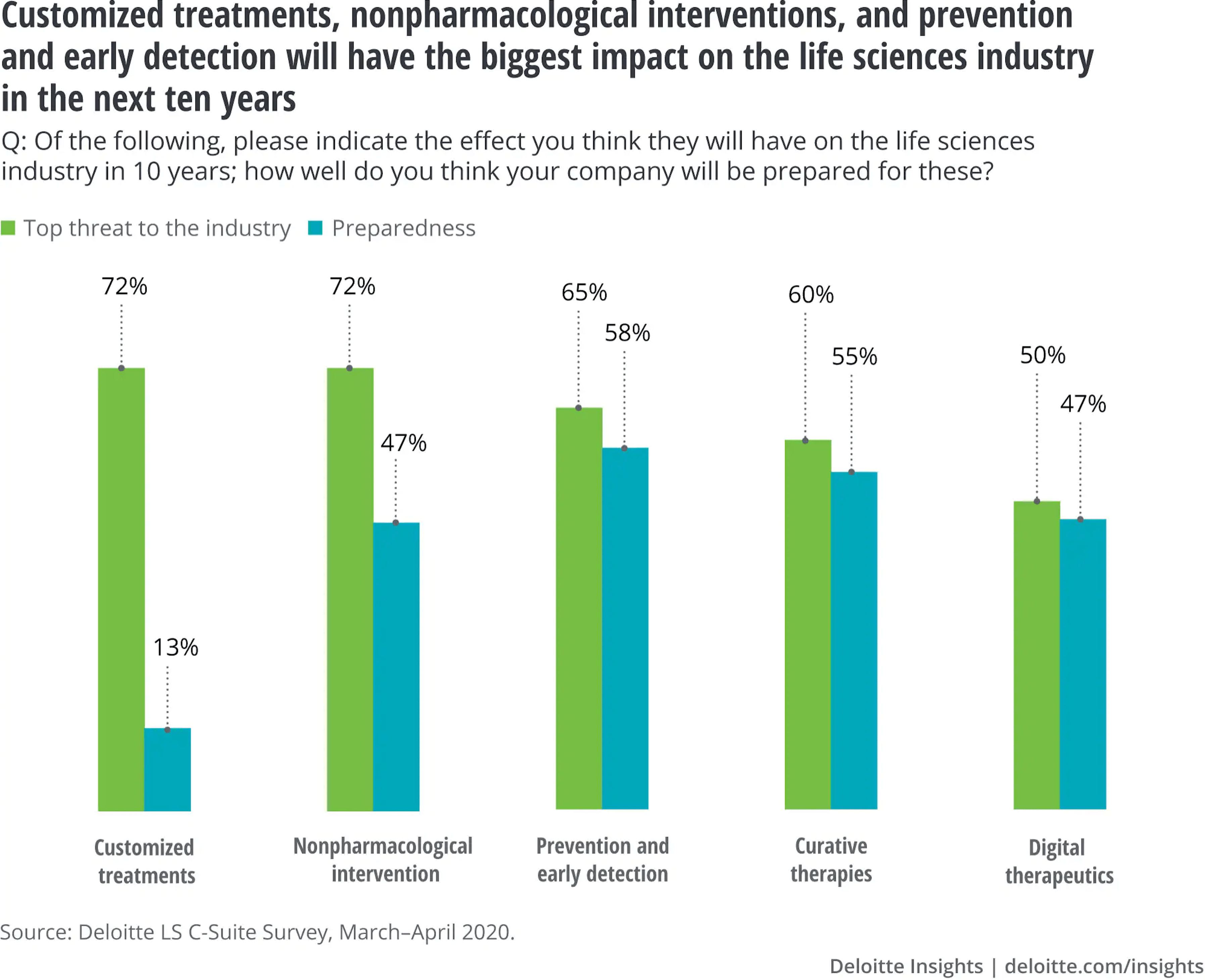
Source: Deloitte Insights
A growing emphasis in healthcare systems on holistic value, outcomes and cost-efficiency is likely to be a prime motivator in steering the industry towards a broader consideration of where it should sit in the evolving healthcare equation. As Maria Whitman, global head of the pharmaceutical and biotechnology practice at consultants ZS points out, value-based care models unsurprisingly put much more emphasis on preventative-care management.
“Breast-cancer screening rates go up, colorectal screening rates go up, cancer screenings, everything you can imagine”, Whitman comments. “So that’s a win-win, but it’s very hard to execute.” The pharmaceutical industry can help here by offering “more than medicine”. It has already transitioned from focusing on product benefits to thinking about driving and improving the patient experience of product utilisation, Whitman points out. The next step, one which a number of companies are already taking, is to engage fully with “an outcomes-focused world”.
That might involve real-world evidence programmes, disease-awareness campaigns, cross-industry partnerships, prediction analytics and broader patient or population screening. Whitman cites real-world initiatives such as Eli Lilly’s TRIUMPH study (Preventive Treatment of Migraine: Outcomes for Patients in Real-World Healthcare Systems), which is evaluating the long-term effectiveness of Emgality (galcanezumab-gnlm) compared with other preventive treatments for migraine.
Another venture into the early care pathway is Novo Nordisk’s sponsorship of Cities Changing Diabetes, a public-private partnership aimed at combating obesity and diabetes in urban environments around the world. The emphasis here is on risk identification, targeted intervention, prevention and health promotion.
As Whitman observes, outcomes-based contracting for complex, high-cost treatments, such as cell and gene therapies, is already nudging pharmaceutical companies away from reactionary business models based on patient numbers and volume sales. Ultimately, though, payment by outcomes or the prevention-oriented programmes cited above are not that radical a departure for an industry already engaged in building a more inclusive ecosystem for treatment uptake.
At the least, though, they show that companies are thinking about the whole patient journey in preventable diseases, and how they can add value beyond stepping in once treatment becomes inevitable. How much this involvement is really in the long-term interests of patients and healthcare systems, or whether it is essentially a Trojan Horse for more nuanced, pragmatic commercial strategies, is open to debate.
"Outcomes-based contracting for complex, high-cost treatments, such as cell and gene therapies, is already nudging pharmaceutical companies away from reactionary business models based on patient numbers and volume sales"
Which is not to say that, in a value-based healthcare environment, industry cannot have it both ways. The question they should be asking, Whitman says, is: “Am I going to stay a medicines provider? Or am I going to expand my breadth to think about playing in that earlier space, and still make money from it without being completely altruistic?”
The road ahead
This blog on pharmaceutical trends for 2023 has described a number of significant challenges and opportunities, some new, and some amplified by exceptional circumstances such as the COVID-19 pandemic as well as cumulative developments like population ageing or healthcare cost inflation.
The disruptive impact and legacy effects of COVID-19 have, among other things, heightened uncertainties over timelines, evidence requirements and outcomes in the HTA, pricing and reimbursement procedures that are increasingly critical component of pharmaceutical market access. The post-pandemic world requires more agile launch execution. Digital tools such as SmartLaunch™ and SmartAccess™ can help to manage these shifting timelines more efficiently, while providing real-time visibility of HTA, reimbursement and launch status and timelines across countries.
Given the rapid pace of change addressed in this pharmaceutical industry analysis 2023, companies need to be all the more confident that their products will be launched and taken up in the marketplace with the maximum available impact. That means optimising digital solutions, and collaboration software specifically, to drive alignment, agility, efficiency, and ultimately a higher quality launch execution.
A 360⸰, real-time view of progress and challenges from market to market will help to ensure that 2023 is the year in which pharma can really start to look beyond the gains, losses and upheavals of the COVID-19 pandemic. Then the industry can reaffirm its focus on a core mission of innovation, growth and continuous improvement in disease management and/or prevention, one that genuinely enhances the lives of patients while helping overburdened healthcare systems to manage their budgets more efficiently and cost-effectively.
