
 Andre Moa
Andre Moa
 17 Nov 2020
17 Nov 2020
 15 minute read
15 minute read

Any fears that COVID-19 might stem the flow of new drug approvals in 2020, depleting or diverting resources at regulatory agencies and their pharmaceutical clients, appear largely premature.
Year-to-date tallies at the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA), the two most influential agencies for global launch programmes, suggest the trend for 2020 new drug approvals is in good health.
Consequently, we can expect another substantial crop of new drug launches during 2021, some of them potential blockbusters. Of course, industry’s launch plans for next year will be coloured by pandemic-related challenges beyond securing regulatory approvals. They range from competition for reimbursement in overstretched healthcare systems to managing virtual sales and marketing strategies, or overcoming supply-chain disruption.
Nonetheless, pharmaceutical companies are not only finding new ways to work remotely but seizing opportunities to address urgent need for COVID-19 treatments, alongside a vaccine for coronavirus. These products could be a significant feature of the launch landscape in years to come.
While all eyes are trained on the more than 200 vaccines now in development for COVID-19 (10 in a Phase 3 clinical trial), 2020 has already seen the first US and EU approvals of a drug treatment specifically for COVID-19, Gilead's Veklury (remdesivir). That was followed in early November 2020 by a US Emergency Use Authorization (EUA: not an actual approval, which remains subject to a successful US Biologics License Application) for Eli Lilly’s monoclonal antibody bamlanivimab to treat mild-to-moderate COVID-19.
FDA and EMA approvals 2020
Keeping those factors in mind, how do FDA and EMA approvals stack up against last year, as we fast approach the end of 2020? Pretty well, as it turns out. The banner figure for the FDA comes from its Center for Drug Evaluation and Research (CDER): 42 new medicines and therapeutic biologics approved by mid-November 2020.
That compared with 38 new medicines/therapeutic biologics approved through the same channel at the equivalent point in 2019, and 48 marketing authorisations granted by CDER for the whole of that year. Progress with new biological agents at the FDA’s Center for Biologics Evaluation and Research (CBER) has been much less encouraging. Just eight of these products had been licensed by mid-November 2020, versus 21 at the same point in 2019 and 22 for the full year.
The EMA has made an impressive showing in the year to date. As of mid-November, the agency’s Committee for Medicinal Products for Human Use (CHMP) had issued positive opinions on 53 new medicines, therapeutic biologics and vaccines. That far outstripped the 39 positive opinions in year-to-date 2019, as well as the 41 positive CHMP recommendations for the full year.
In most cases, the European Commission follows the CHMP’s positive opinions by granting final approval to market a new medicine EU-wide. There may be several months’ lag between CHMP recommendation and Commission sign-off, however. At the time of writing, 34 of the 53 new products recommended by the CHMP for EMA approval had secured marketing authorisations from the Commission, according to publicly available information. Several more EU approvals were expected during the fourth quarter of 2020.
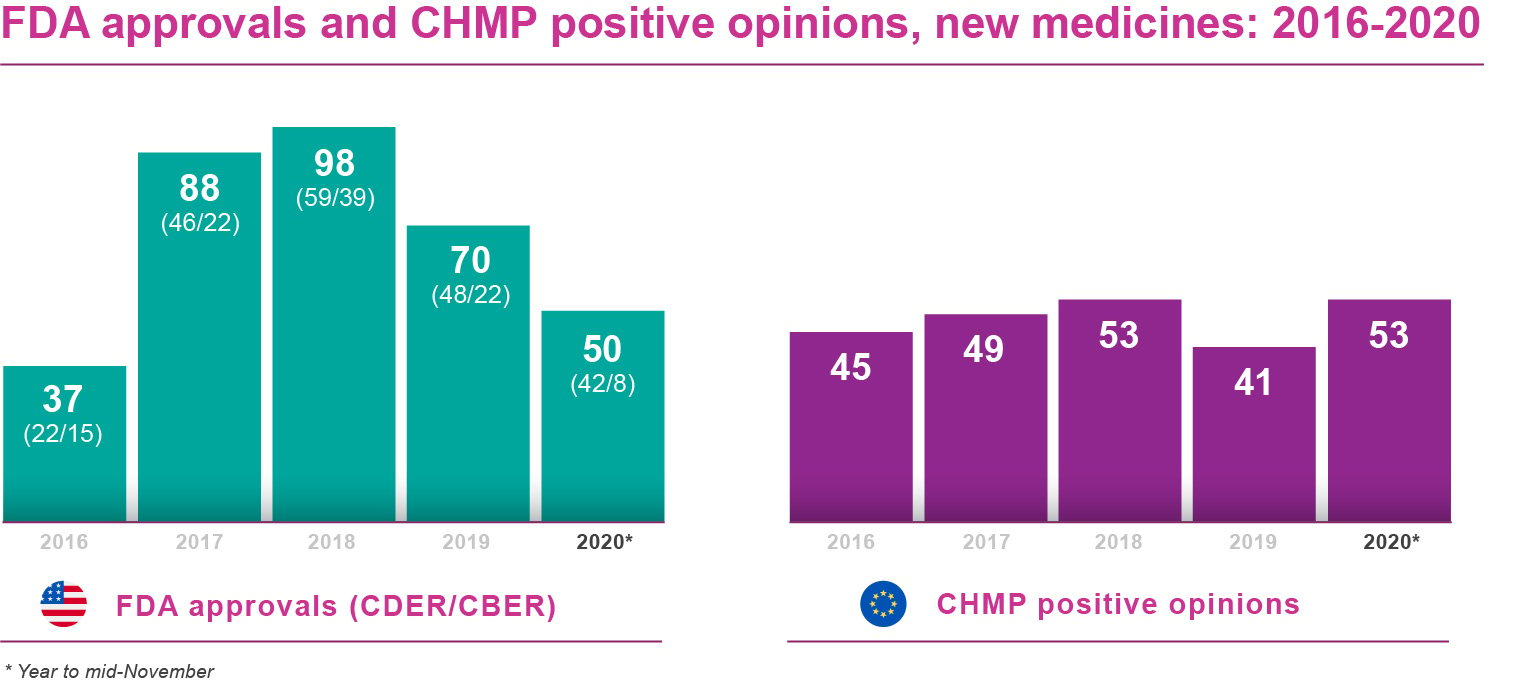
As the table indicates, 2018 was an exceptional year for new drug approvals on both sides of the Atlantic, although the CHMP’s positive opinions in 2020 to date are already level its 2018 tally. Within the FDA, it would be a considerable stretch for the CBER to get anywhere near the 39 biological agents it OK’d in 2018, although this year’s approvals may be boosted (at least symbolically) by one or two EUAs for COVID-19 vaccines in December.
|
New and novel medicines
For the purposes of this review, FDA approvals omit generics and biosimilars. The CHMP recommendations here do not include generics or biosimilars either, nor products approved through hybrid or informed consent procedures that rely on pre-existing data from other medicines or biologics.
For FDA registration, the CDER lists ‘novel medicines’, which are typically, but not exclusively, new molecular entities or NMEs and can extend to diagnostic agents. CBER approvals include gene/cell therapies and vaccines, but many of them are for screening tests, assays or reagents.
EMA-approved ‘new medicines’ include chemically based drugs as well as biologicals and vaccines. Whether products awaiting final EMA registration are actually ‘new active substances’ – the nearest equivalent to FDA-approved NMEs – is not clear until the European Commission gives its approval and the relevant documentation appears on the EU’s website. |
Drug-approval numbers can wax and wane from year to year for a number of reasons, not necessarily all related to available resources and expertise or the complexity of the applications under review. There may, for example, be significant spill-overs from late in one year to early in the next, fluctuations in the quality and timing of submissions, or unique disruptions such as COVID-19 or the EMA’s Brexit-related relocation from London to Amsterdam during 2019 and early 2020.
Of course, marketing authorisation does not guarantee launch, at least not in all available markets, let alone launch excellence. Many variables remain, such as local market conditions, pricing, reimbursement and, increasingly, health technology assessment (HTA). This is where launch planning comes in. Barring unexpected setbacks, companies with an FDA approval and/or CHMP recommendation will want to access as many key global markets as possible, with optimal timing and impact.
Breakthrough innovation
Volume apart, what do this year’s approval counts tell us? Breakthrough innovation, especially for rare diseases, continues to play a defining role in US and EU approval trends. Other than Veklury, regarded by sceptics as a blockbuster-in-waiting that offers at-best modest efficacy, the approval lists include gene and cell therapies such as:
- Orchard Therapeutics’ Libmeldy (cryopreserved autologous CD34+ cells encoding the arylsulfatase-A gene) for metachromatic leukodystrophy; and
- Gilead's Tecartus (autologous anti-CD19-transduced CD3+ cells) for mantle cell lymphoma.
Among other notable approvals/recommendations were, in the EU:
-
Givlaari(givosiran), the first treatment for the rare genetic disorder acute hepatic porphyria (the FDA OK’d Alnylam Pharmaceuticals’ drug in November 2019);
- Novo Nordisk’s Rybelsus(semaglutide, FDA-approved in September 2019), the first oral GLP-1 receptor agonist for type 2 diabetes;
- AveXis/Novartis’ gene therapy Zolgensma(onasemnogene abeparvovec, FDA-approved in May 2019) for the treatment of spinal muscular atrophy.
In all, 21 of 53 products recommended for approval by the CHMP in the year to date (10 out of 39 in the same period of 2019) were orphan medicines, underlining the continuing shift to specialty drugs in pharma.
A number of US approvals so far in 2020 were also under orphan-drug designations, such as:
- Roche’s Enspryng (satralizumab-mwge) for neuromyelitis optica spectrum disorder;
- Regeneron’s Inmazeb (atoltivimab, maftivimab, odesivimab-ebgn), the first FDA-approved treatment for Ebola virus;
- NS Pharma’s Viltepso (viltolarsen) for gene-mutated Duchenne muscular dystrophy;
- Incyte’s Pemazyre (pemigatinib), the first FDA-approved treatment for advanced or metastatic FGFR2-fusion bile-duct cancer; and
- Horizon Therapeutics’ Tepezza (teprotumumab-trbw), the first approved drug for thyroid eye disease.
Among other innovative medicines securing US approvals in 2020 were:
- Immunomedics’ Trodelvy (sacituzumab govitecan-hziy) for metastatic triple-negative breast cancer; and
- Aimmune’s first-in-class peanut-allergy immunotherapy Palforzia (Arachis hypogaea Allergen Powder-dnfp).
Blockbuster potential
For companies with innovation to sell, pharmaceutical launch success inevitably comes down to revenues, profit margins and lifecycle management. Moreover, innovation only has meaning if it reaches patients in need.
Among new drugs launched or close to market in 2020, commentators have identified a number of potential blockbusters. These include:
- Novo Nordisk’s Rybelsus;
- Bristol-Myers Squibb’s Zeposia (ozanimod) for relapsing multiple sclerosis;
- AstraZeneca/Daiichi Sankyo's Enhertu (fam-trastuzumab deruxtecan-nxki) for HER2-positive breast cancer; and
- Novartis/The Medicines Company’s siRNA PCSK9 inhibitor Leqvio(inclisiran) for primary hypercholesterolaemia or mixed dyslipidaemia.
Cortellis’ (Clarivate Analytics) Drugs to Watch report for 2020 lists eleven drugs set to enter the market this year and achieve blockbuster status (annual sales of US$1 billion or more) by 2024. Not all of these prospects will be peaking anytime soon.
For example, both BioMarin’s haemophilia A gene therapy valoctocogene roxaparvovec (Valrox) and Gilead's selective JAK-1 inhibitor for rheumatoid arthritis, Jyseleca (filgotinib), have been hit by FDA requests for additional data that could substantially delay US approval. Jyseleca already has regulatory clearance in the EU and Japan, whereas the European Medicines Agency has also asked for more clinical-trial data on Valrox.
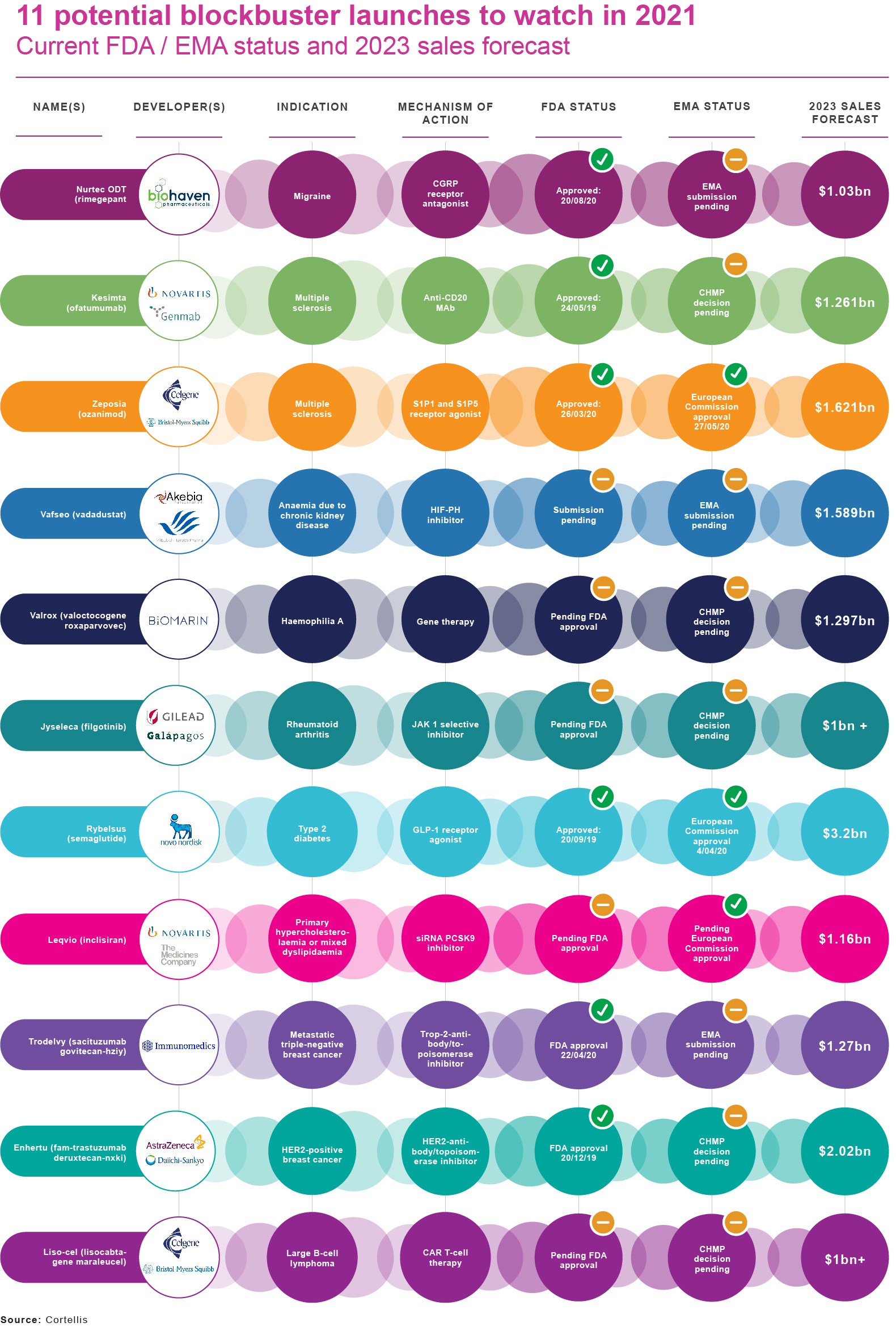
As Clarivate Analytics points out, nearly half of the 11 products in this year’s Drugs to Watch list are biologics; almost all of them benefited from accelerated-development pathways. They racked up 18 orphan-drug designations in total, while four products were classified as breakthrough therapies in the US and two earned EU PRIME (priority medicines) status.
From R&D to launch excellence
In the meantime, drug-development pipelines just keep on getting fatter. According to Informa’s annual Pharmaprojects Pharma R&D review, there were 17,737 pipeline projects ongoing in early 2020, 9.6% more than in the previous year’s analysis, and outpacing pipeline growth rates of 6.0% in 2019 and 2.7% in 2018.
As Pharmaprojects Senior Director Ian Lloyd observes, though, such fecundity can turn into costly bloat unless sufficient numbers of drug candidates achieve approval and market access. That calls for the right launch management tools to ensure that milestones, timelines and challenges in pharmaceutical launch plans are visible, co-ordinated and actionable in real time across different countries, functional silos and layers of management.
Technology-enabled cross-functional alignment and collaboration, across the whole spectrum of activities driving launch readiness and excellence, raise the odds that continuing buoyancy in drug approvals will translate into new growth for industry, and new opportunities for patients to manage or even conquer their disease.
TRiBECA® Knowledge provides pioneering SaaS platforms to some of the world’s leading pharmaceutical companies, helping them to embrace digital transformation and optimise their product launch, market access and tendering operations. Our collaborative launch readiness platform SmartLaunch™ raises the odds of converting successful approvals into launch excellence by enhancing visibility, alignment, transparency, communication and collaboration across products, management layers, business functions and countries worldwide. That goes not only for newly approved novel therapies but for the new indications and line extensions that remain the backbone of pharmaceutical product lifecycle management.